
随着高通量测序技术的发展,转录组测序已经成为研究基因表达调控的主要手段。二代测序技术通量高,更多的关注基因表达量,然其局限性在于测序读长短,转录本产生拼接错误,导致转录本结构不完整。生物体内复杂多变的转录本是调节基因表达和蛋白质多样性的重要机制,准确鉴定转录本结构,是深入研究基因表达调控模式的基础。
全长转录组研究可全面快速的获取有参或无参物种特定组织或器官在某一状态下的全长转录本信息,构建转录本基因集。相较于RNA-seq,基于Nanopore三代测序平台的转录组研究,无需打断,直接读取5’ 端到3’-PolyA的高质量完整转录本,准确鉴定可变剪接、新基因/新异构体、可变多聚腺苷酸化、融合基因等,完善基因组注释。此外,Nanopore平台更拥有Direct RNA测序方式。
那么,使用Direct RNA测序技术,我们能做些什么呢?下面小编带大家解析一篇新出炉的direc RNA 测序文章
中文题目:使用direct RNA测序技术对秀丽杆线虫进行转录组测序
英文题目:The full-length transcriptome of C. elegans using direct RNA sequencing
发表时间:2019.04.09
发表期刊:BioRxiv
研究背景
目前绝大多数的转录组注释都是依赖于cDNA高通量测序固有的短读长测序技术。线虫的基因组紧凑,注释良好,细胞谱系稳定,是一种理想的实验模型生物。然而,在秀丽杆线虫的转录组中,大约有超过一半的转录本缺少全长信息的支撑,且依赖于无法跨越转录本全长序列的短reads的预测。同时,利用短读长对polyA和3’UTR进行预测,并不能直接鉴定到剪接转录本的起始位点,且在3’UTR鉴定中,依赖于将推测的剪接位点分配给最近的重叠或上游基因。相比之下,纳米孔测序没有理论上的读取长度上限,能够在单个分子水平上对转录本进行一端到另一端的测序。
材料方法
材料:L1、L2、L3、L4、幼年成虫(YA)、成虫,雄性线虫;每个时期两次技术重复
方法:20 µg Total RNA,调取约600ng PolyA RNA;GridION平台,direct RNA sequencing
结果
1、线虫测序数据统计及全长转录本鉴定
选取线虫发育的L1、L2、L3、L4、幼年成虫(YA)和成虫时期,以及雄性成虫,其中幼年成虫和成虫雌雄同体,对其进行direct RNA测序,每个时期两个技术重复。共计获得5.54M reads,其平均长度为739到934 bp,基因组比对率为87.8%。
通过一系列的筛选标准,最终共计获得2.9M 全长reads。综合所有阶段,最终鉴定到25,944 个全长转录本,其中20,987个转录本有唯一的可变剪接形式,16,325个转录本有唯一的3’UTRs ,平均每个阶段鉴定到超过12,000条全长序列。在和线虫数据库进行注释比对后,有12,613 转录本和10,711 个基因有全长数据支持,此外还鉴定到4,234个新基因和7,404 个新转录本。其中,发现9,900个已知可变剪接转录本和2,188个新的可变剪接转录本,对应1,349个基因。在这些新的剪接转录本中,有1,283个转录本在注释的供体和受体剪接位点之间存在新的剪接位点,同时173个转录本还存在新的外显子。
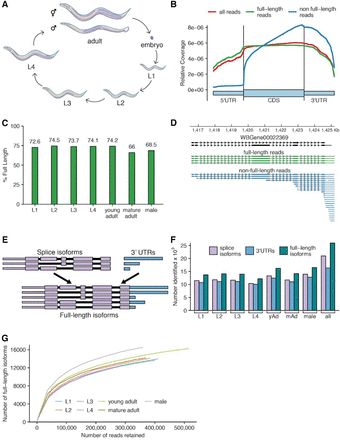
图一 全长转录本测序概述
2、3’UTR鉴定
本文共鉴定到16,325个唯一的3’UTR转录本,在每个阶段均鉴定到超过10,000 个3’UTRs。将鉴定到的3’ UTR与已知数据库进行比对,发现共有82.9% UTRs的重叠。此外,还鉴定到2,304个新的发现的3’ UTR。3’ UTR的长度会随着阶段的延续,从L1到L4,逐渐变短,在成年线虫中,雄性线虫的3’ UTR要短于雌雄同体的成虫,而成虫的 3’ UTR要稍长于L4阶段的成虫,这与前人的报道相反。通过不同阶段多聚腺苷酸化位点统计,发现在不同阶段,其位点不存在显著差异,该结果表明,不同阶段3’ UTR的长度分布与多聚腺苷化位点无关。
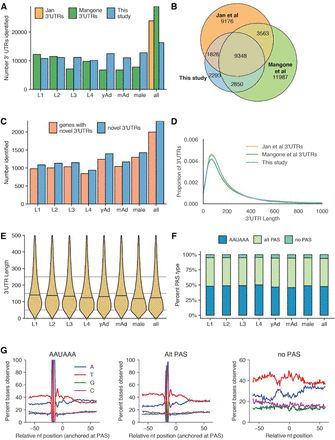
图二 3’ UTR特征统计
3、PolyA尾预测
研究表明,在黑腹果蝇中,polyA尾的长度会随着发育阶段而呈现出动态变化。而在本文中,通过对线虫不同时期polyA长度的统计,发现其变化比较稳定,在幼虫发育阶段,其变化范围为49nt(L1)到54nt(L2);在成虫发育阶段(幼年成虫、雌雄同体成虫和雄性成虫),其polyA长度中位数为58nt,要长于幼虫的polyA长度(52nt)。该结果表明,在成虫和幼虫之间,polyA的长度变化最为显著。将L4阶段的polyA长度分布与前人研究报道进行比较,发现其长度分布非常相似,均在30~40 nt出现峰值,并向较长的尾部延伸。
PolyA的长度,有可能和3’ UTR区域的polyA剪接位点(AAUAAA)有关。为了证明该推测,本文对不同polyA剪接位点类型(经典的、可变的及无剪接位点)的polyA尾进行了长度统计,发现不同类型间存在显著差异,且3’UTR区域不存在polyA剪接位点的,具有更长的polyA尾巴。在3’UTR区域,无polyA剪接位点的,其polyA长度中位数为58 nt,具有可变polyA剪接位点的,长度为46 nt;具有经典的polyA剪接位点(AAUAAA),其长度中位数为48 nt。研究表明,polyA尾的长度与基因表达呈现负相关,即高表达的基因具有更短的polyA尾。对本研究数据进行统计发现,在幼虫发育阶段发现有相似的负相关关系。例如,Y37E3.8基因的a转录本的表达要显著高于b.1转录本,其polyA尾相较于b.1更短。polyA长度和基因表达水平的相关性R2 最高为0.1297。在成虫发育阶段,polyA长度和基因表达水平的相关性并不是很高。该结果表明,基因表达水平和polyA尾长度的负相关性具有阶段性。最后,本文研究了polyA长度与内含子保留可变剪接事件的相关性。研究表明,在人类细胞系中,polyA尾的长度与内含子保留有关。在本文中,也发现polyA尾和内含子保留事件呈现正相关。该结果表明,polyA尾在转录后调控存在一种保守机制,即在核转录本中,其拥有更长的polyA尾,而随着转移到内质网的过程中,polyA尾会进行脱腺苷化而进行转录后进程。
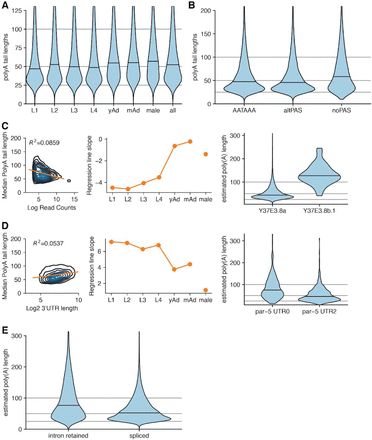
图三 PolyA 特征统计
小结
在本研究中,作者更侧重于关注全长转录本的结构分布,并对其进行了详细描述。从本研究可以看出,使用Nanopore测序平台进行direct RNA测序,借助其长读长优势,可准确鉴定转录本的结构信息。此外,Nanopore平台还拥有cDNA测序方式(橄榄果蝇胚胎发育的动态变化解析),多种建库方式,可满足不同的研究需求。




 京公网安备 11011302003368号
京公网安备 11011302003368号