

现代作物遗传改良依赖于农艺性状遗传与分子机制的解析,大部分农艺性状属于数量性状,并由复杂的遗传网络调控。数量性状基因(QTGs)的克隆对于作物改良极为重要,传统方法是通过图位克隆定位基因,然而此法需要构建高世代群体,大群体筛选交换单株以及考察详尽的田间表型,因此,比较费时、费力、费钱。虽然关联分析也是解析QTGs的有效方法,但是很难检测到稀有等位基因(通常优异农艺性状所具有的),并且建立群体大小适宜与品种多样性丰富的种质资源平台需要耗费较大的代价。
被广泛应用于遗传基因定位的另一种方法,BSA,也可有效解析质量与数量性状。高通量测序技术的快速发展使BSA在基因定位中发挥着巨大潜力,例如,MutMap,SHOREmap与MMAPPR在质量性状定位中可取代传统的图位克隆。对于数量性状,可利用QTL-seq定位QTLs,但定位区间较大很难识别候选基因,所以非常需要一种新的方法,用于快速定位数量性状候选基因。
介于上述种种定位方法的局限性,本文介绍一种新的方法——QTG-Seq,即通过QTL分离(即,将数量性状转化为质量性状),极端表型混池,高通量测序以及新算法挖掘候选基因。这一方法是由华中农业大学李林课题组、章元明课题组与中国农科院王国英课题组开发并应用成功,最后以“QTG-seq accelerates QTL fine mapping through QTL partitioning and whole-genome sequencing on bulked segregant samples”为题,在线发表在Molecular Plant上。

QTG-Seq基本原理(流程如下)——
(1)群体构建: F1, F2, BC1F1群体
(2)QTL定位:F2群体定位QTL,F2:3家系证明QTL定位结果
(3)QTL分离:分子标记筛选目标QTL杂合,其它QTL纯合的BC1F1单株(BC1F1数目要充足);然后自交得到BC1F1:2家系
(4)极端表型选择:从BC1F1:2中选2个极端表型组(前20%,后20%,至少1000个单株)
(5)极端表型混池:提取DNA并混池,形成2个极端池
(6)测序并变异calling:对两个混池测序,与参考基因组比对分析变异
(7)关联分析:新方法smoothLOD(准确关联靶标位点),结合其他方法,如ED(Hill et al., 2013)与G’(Magwene et al., 2011)(关联peak位置)

图1. QTG-Seq基本原理
应用——QTG-Seq快速定位玉米株高QTL,qPH7
以玉米株高为模式性状展开QTG-seq研究:利用4代玉米群体材料,通过QTL-seq精细定位株高主效QTL,这与传统QTL精细定位用到许多代创制高世代回交群体明显不同。首先,利用株高差异明显的两个自交系HZS与1462,构建F2分离群体,个体株高变化范围为199cm~307 cm,呈超亲分离的现象,暗示株高是由多个QTLs控制;然后,利用1028个多态性标记定位主效QTLs,结果定位到4个QTL(位于Chr.1,3,6,7),PVE介于7%-17%,又利用F2:3家系验证QTL定位结果可靠;最后,选择Chr7.上的QTL qPH7利用QTL-seq策略进行精细定位。
利用12个标记筛选出813个BC1F1单株(qPH7位点杂合,另外3个QTL位点纯合),从中选择15个材料,自交获得BC1F1:2家系,并考察株高。共获得3120个BC1F1:2单株,从中划分出2个极端表型组,低值组580个单株,高值组567个单株,并分别组成混池(低池与高池);接着,对混池展开全基因组测序(>280×),检测到197021个高质量SNPs。利用smoothLOD与ED4算法分析出Chr.7只有一个峰,并且峰的位置在135.3Mb。KASP标记分型也证实峰的位置处于135.1与135.2之间。有趣的是,smoothLOD法将qPH7定位在chr:135216475bp,此位点正好处在Zm00001d020874上。Zm00001d020874编码一个NF-YC结构域蛋白,它的拟南芥同源蛋白与开花时间有关。
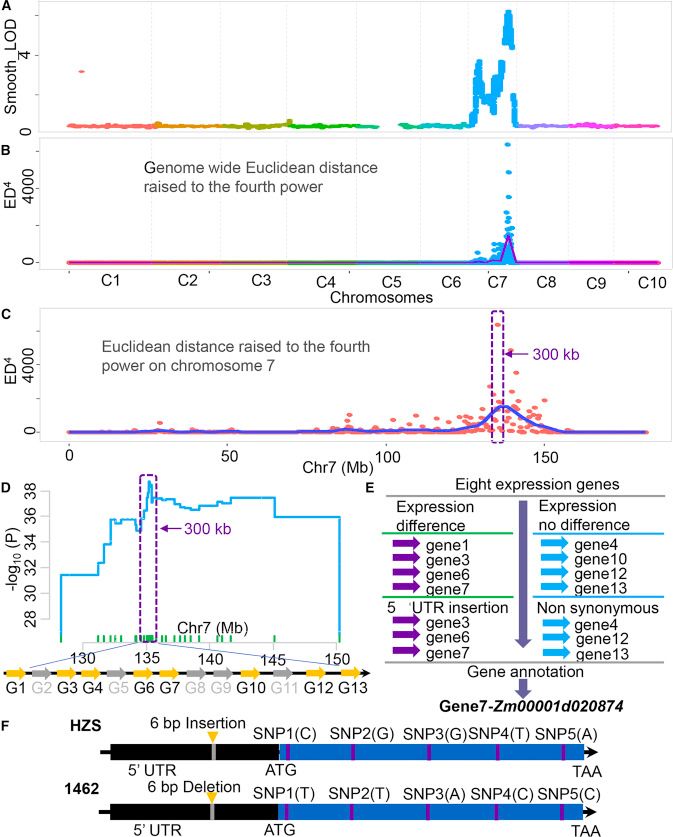
图2. qPH7精细定位与候选基因挖掘
虑到分离群体中重组blocks的存在,利用增加滑窗大小的方法识别目标区域的重组blocks。由于smoothLOD与G’为连续变量,所以采用ED/SNP的方法分析重组blocks,结果显示候选基因与peak间距离<150kb。因此,候选基因的区间在300 kb,其中包含13个基因,也包括基因Zm00001d020874。
因为交换单株数量与定位区间大小密切相关,所以确保较大的混池规模至关重要。根据模拟数据,QTG-seq相对低的测序深度(<100×)足够检测出所有的重组blocks,因为每个重组block包含许多SNPs,没必要对每个重组block的所有reads测序。因此,为了平衡定位精度与测序成本,在使用QTG-seq方法时,建议增大混池规模与相对低的测序深度。
为进一步确定候选基因,本研究又通过RNA-seq分析发现,Zm00001d020874表达量在双亲的茎顶端分生组织(SAM)与幼嫩节间组织中存在显著性差异。基于拟南芥同源蛋白功能与smoothLOD定位信号,认为Zm00001d020874是qPH7的候选基因,并且后续的CRISPR基因敲除,蛋白互作验证等实验均暗示Zm00001d020874是控制株高的基因。
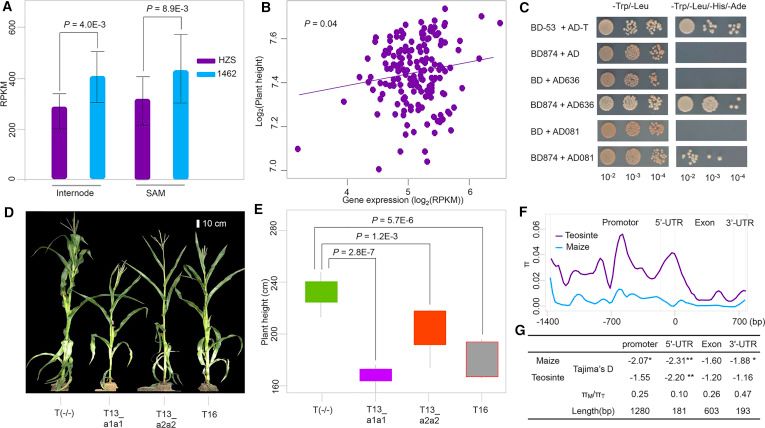
图3. 候选基因的验证
Monte Carlo模拟支持QTG-Seq的可靠性
4个Monte Carlo模拟实验如下:
1. PVE与QTG位置偏差大小:5%时,接近80kb;≥10%时,30-60kb
2. 取样率与QTG位置偏差大小:当取样率为20%时,位置偏差略大;较大的取样比例会增加群体大小,但有效重组事件数量也会增加
3. 群体大小与QTG位置偏差大小:当群体大于4000时,位置偏差小于60kb
4. 测序深度与检测力:当测序深度大于100×时(混池超过1000个个体),检测力达到100%
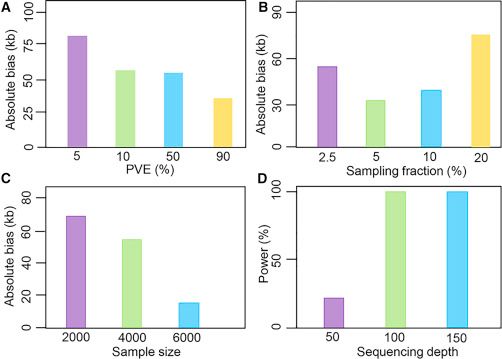
图4. SmoothLOD算法影响定位准确性的各种因素
QTG-Seq特点总结:
1. 省时省力省钱,不需要构建高世代复杂群体,只需F1, F2, BC1F1群体即可实现;
2. 将多个QTL分解为单个QTL分析(数量性状→质量性状),即,在每个QTL区域选择3个(或多个)多态性标记筛选目标QTL杂合,其它QTL纯合的交换单株,确保交换单株后续自交发生表型分离以对目标QTL进行解析;
3. 测序策略:高的混池个体数目(数百至上千个个体)与相对低的测序深度(~100×);相对低的测序深度,是因为每条重组block上有多个SNPs,检测其中1个SNP就可代表;
4. 关联分析采用新方法smoothLOD,并结合ED与G’,定位精度高,可识别候选基因;smoothLOD可准确关联靶标位点,而ED与G’关联peak位置;
5. 可结合KASP标记分型寻找重组断点等,识别候选基因。
参考文献:
Zhang H., Wang X., Pan Q., Li P., Liu Y., Lu X., Zhong W., Li M., Han L., Li J., Wang P., Li D., Liu Y., Li Q., Yang F., Zhang Y.-M., Wang G., and Li L. QTG-Seq Accelerates QTL Fine Mapping through QTL Partitioning and Whole-Genome Sequencing of Bulked Segregant Samples. Mol Plant. 2019 Mar 4;12(3):426-437.
如果您的科研项目有问题,欢迎点击下方按钮咨询我们,我们将免费为您设计文章方案。





 京公网安备 11011302003368号
京公网安备 11011302003368号