
是不是会经常绘制热图?那热图怎么做?先看一眼这个函数的参数,这么多,而且最后还有省略号。那么我们应该怎么合理使用这些参数让你的热图看起来更加高大上呢?
pheatmap(mat, color = colorRampPalette(rev(brewer.pal(n = 7, name =
“RdYlBu”)))(100), kmeans_k = NA, breaks = NA, border_color = “grey60”,
cellwidth = NA, cellheight = NA, scale = “none”, cluster_rows = TRUE,
cluster_cols = TRUE, clustering_distance_rows = “euclidean”,
clustering_distance_cols = “euclidean”, clustering_method = “complete”,
clustering_callback = identity2, cutree_rows = NA, cutree_cols = NA,
treeheight_row = ifelse((class(cluster_rows) == “hclust”) || cluster_rows,
50, 0), treeheight_col = ifelse((class(cluster_cols) == “hclust”) ||
cluster_cols, 50, 0), legend = TRUE, legend_breaks = NA,
legend_labels = NA, annotation_row = NA, annotation_col = NA,
annotation = NA, annotation_colors = NA, annotation_legend = TRUE,
annotation_names_row = TRUE, annotation_names_col = TRUE,
drop_levels = TRUE, show_rownames = T, show_colnames = T, main = NA,
fontsize = 10, fontsize_row = fontsize, fontsize_col = fontsize,
display_numbers = F, number_format = “%.2f”, number_color = “grey30”,
fontsize_number = 0.8 * fontsize, gaps_row = NULL, gaps_col = NULL,
labels_row = NULL, labels_col = NULL, filename = NA, width = NA,
height = NA, silent = FALSE, na_col = “#DDDDDD”, …)
第1步:数据准备
用来画热图的数据
- mat:用来画热图的数据参数,一般是一个矩阵,数据是基因表达值,行代表基因,列代表样本。
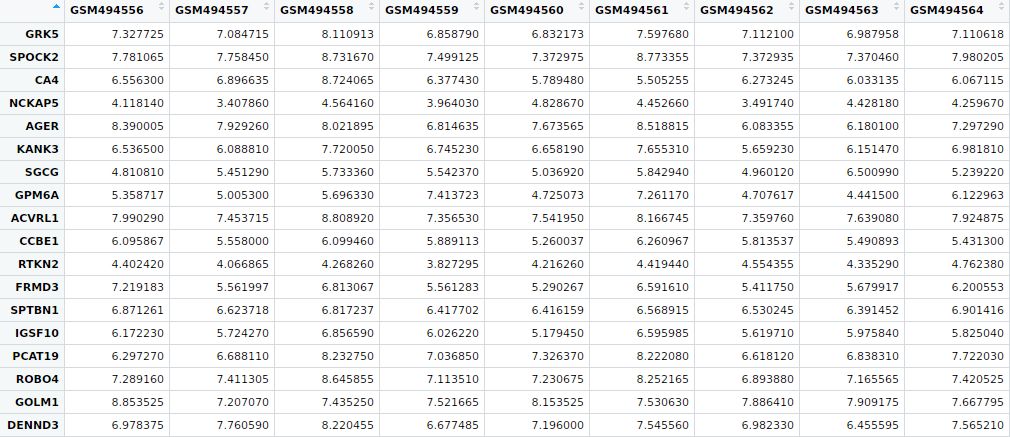
此次例子,我们选择了一套GEO数据库的肺癌数据,数据编号为GSE19804,120个样本,其中包含60个癌症样本和60个癌旁正常样本,前面我们使用t检验,并对p值进行BH校正,筛选fdr小于0.01的基因中前100个在癌症相对于正常样本中显著差异表达的基因进行热图绘制。
部分数据显示如下:
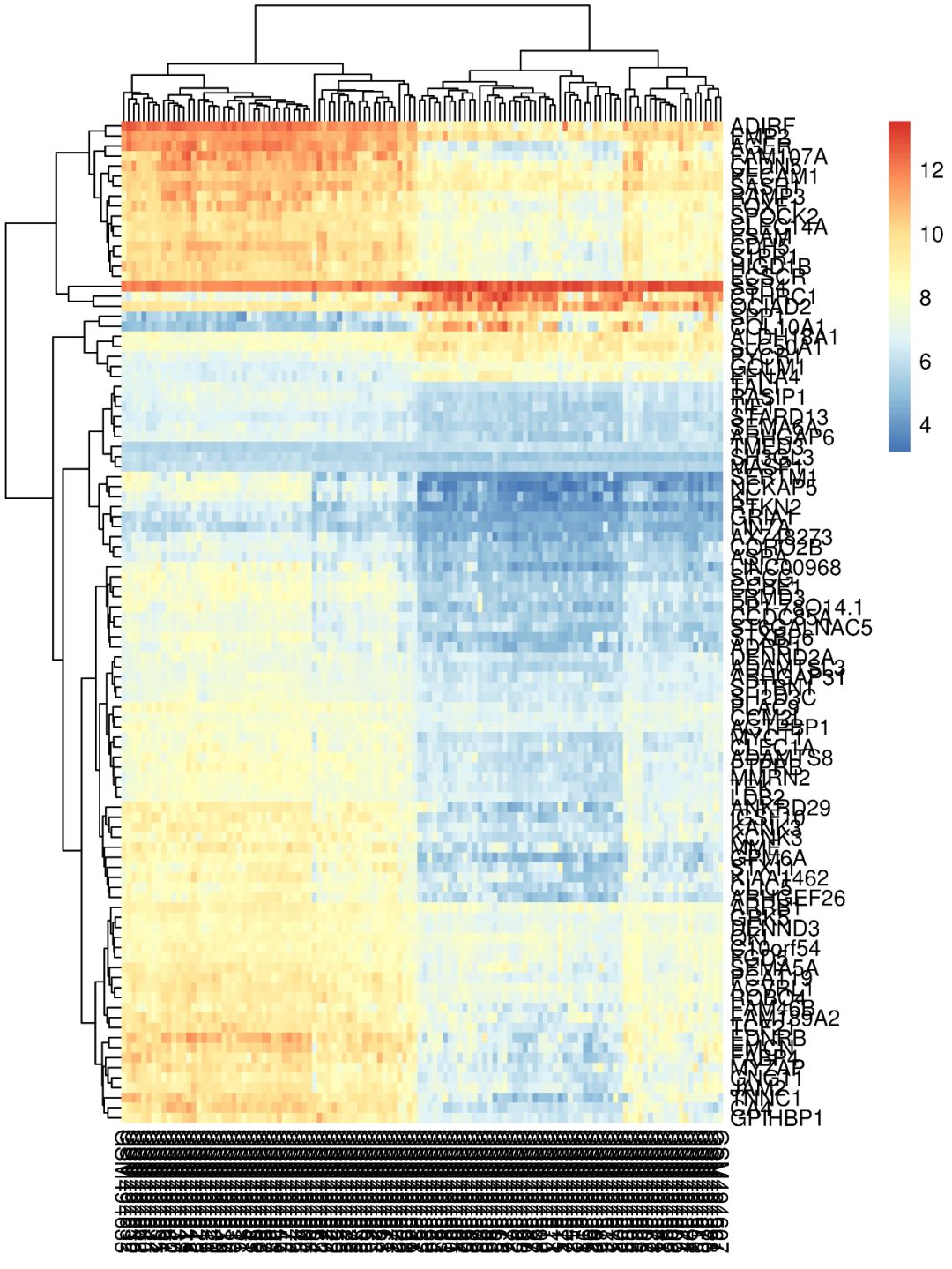
第2步:画图
如使用默认参数画出来的热图,是不是很不好看?基因名和样本名乱成一堆,也看不出来那些样本聚类到了一起…这类热图怎么做?

第3步:参数调整
颜色参数
- color:表示颜色,用来画热图的颜色,可以自己定义,默认值为colorRampPalette(rev(brewer.pal(n = 7, name =”RdYlBu”)))(100),RdYlBu也就是Rd红色,Yi黄色,Bu蓝色的过度,则主调色为红黄蓝。
我们现在将其色调改为红白蓝,看下图,是不是立马好看多了?这种热图不知道该选什么颜色?网址颜色大全https://www.color-hex.com/color-names.html送给你。

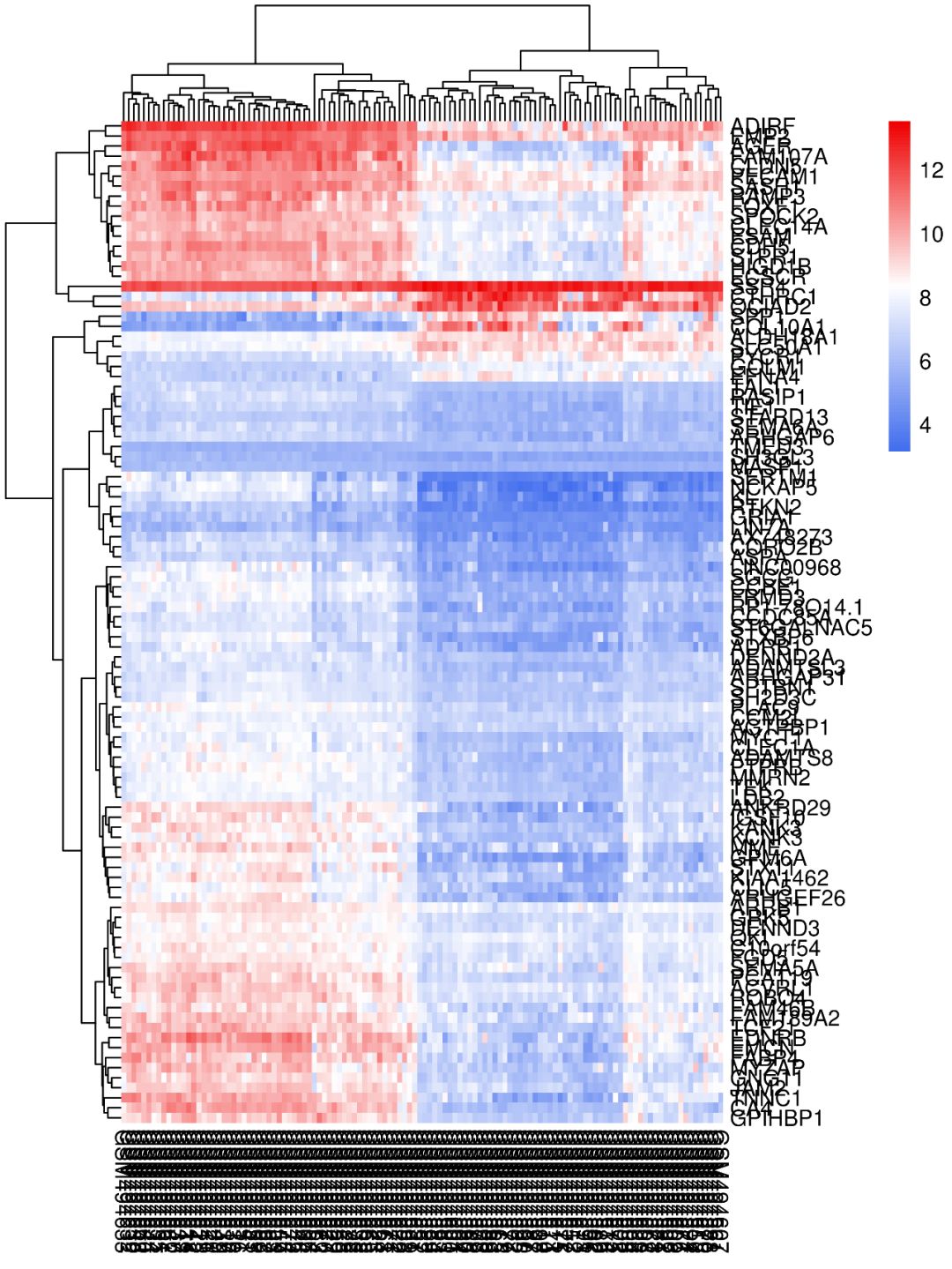
数据变换参数
- scale:是指对数值进行均一化处理,在基因表达量的数据中,有些基因表达量极低,有些基因表达量极高,因此把每个基因在不同处理和重复中的数据转换为平均值为0,方差为1的数据,可以看出每个基因在某个处理和重复中表达量是高还是低,一般选择做row均一化。
- clustering_method:表示聚类方法,值可以是hclust的任何一种,如”ward.D”,”single”, “complete”, “average”, “mcquitty”, “median”, “centroid”, “ward.D2″。
- cluster_rows:表示行是否聚类,值可以是FALSE或TRUE
- clustering_distance_rows:行距离度量的方法,如欧氏距离
- cutree_rows:行聚类数
- treeheight_row:行聚类树的高度,默认为50
- gaps_row:对行进行分割,就不应对相应的行进行聚类
- cluster_cols:表示列是否聚类,值可以是FALSE或TRUE
- clustering_distance_cols:列距离度量的方法
- cutree_cols:列聚类数
- treeheight_col:列聚类树的高度,默认为50
- gaps_col:对列进行分割,就不应对相应的列进行聚类
我们现在将数据进行行均一化,聚类方法选择complete,进行均一化之后的热图还没有前面的好看,查看数据表达情况,我们发现最小的表达值3.149和最大的表达值13.533之间并没有相差很大,因此这套数据其实不适合做均一化处理,画出来的热图更好看。
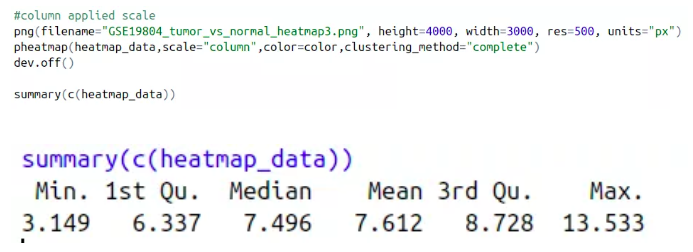
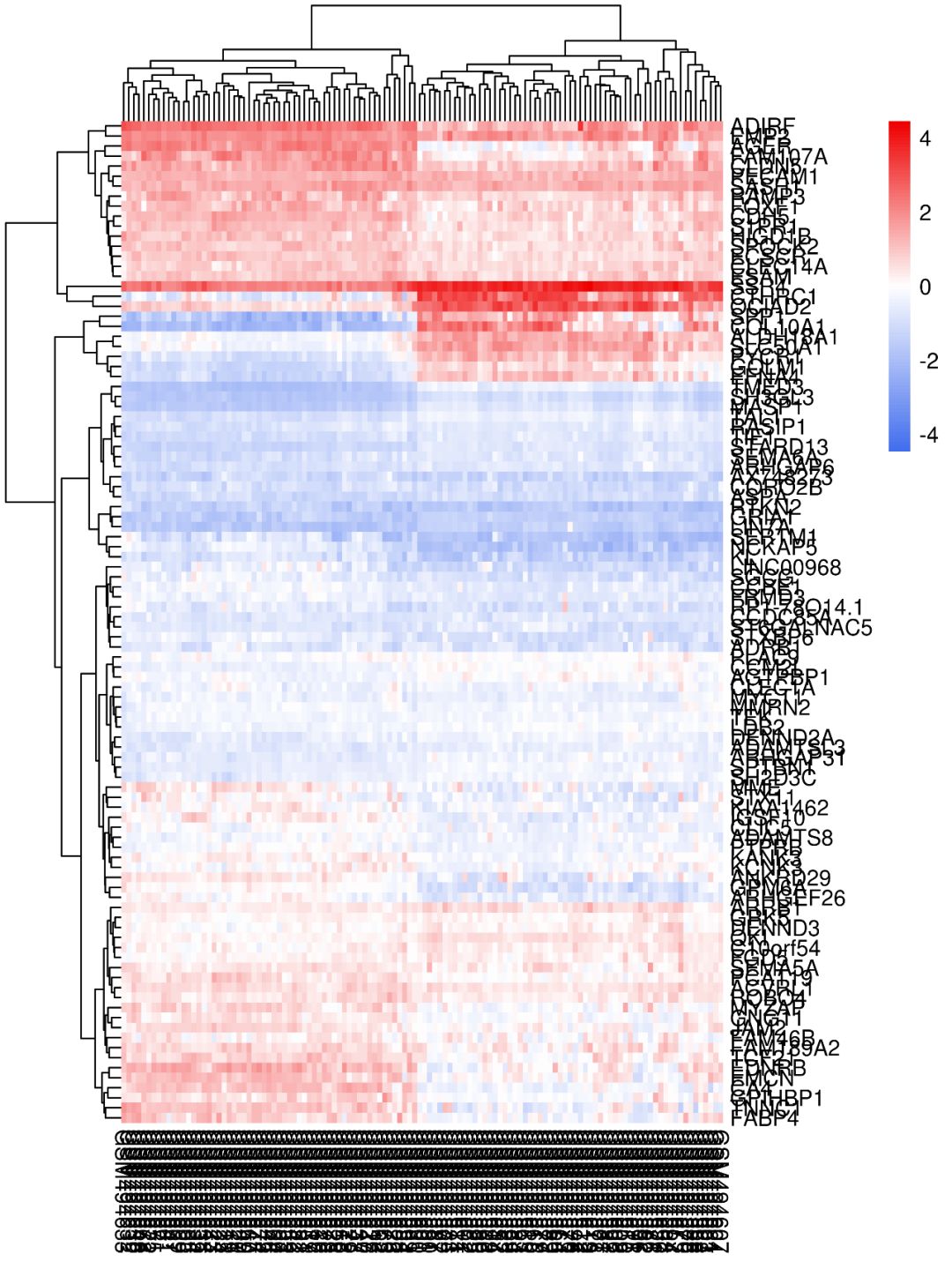
色度条
就是热图右上角那个小小的长方条
- legend:逻辑值,是否显示色度条,默认为T
- legend_breaks:显示多少个颜色数值段
- legend_labels:对色度条上对应位置的字符进行修改
我们对其进行字符修改,可以看到色度条上相应的数字变成了表示颜色高低表达的字符。

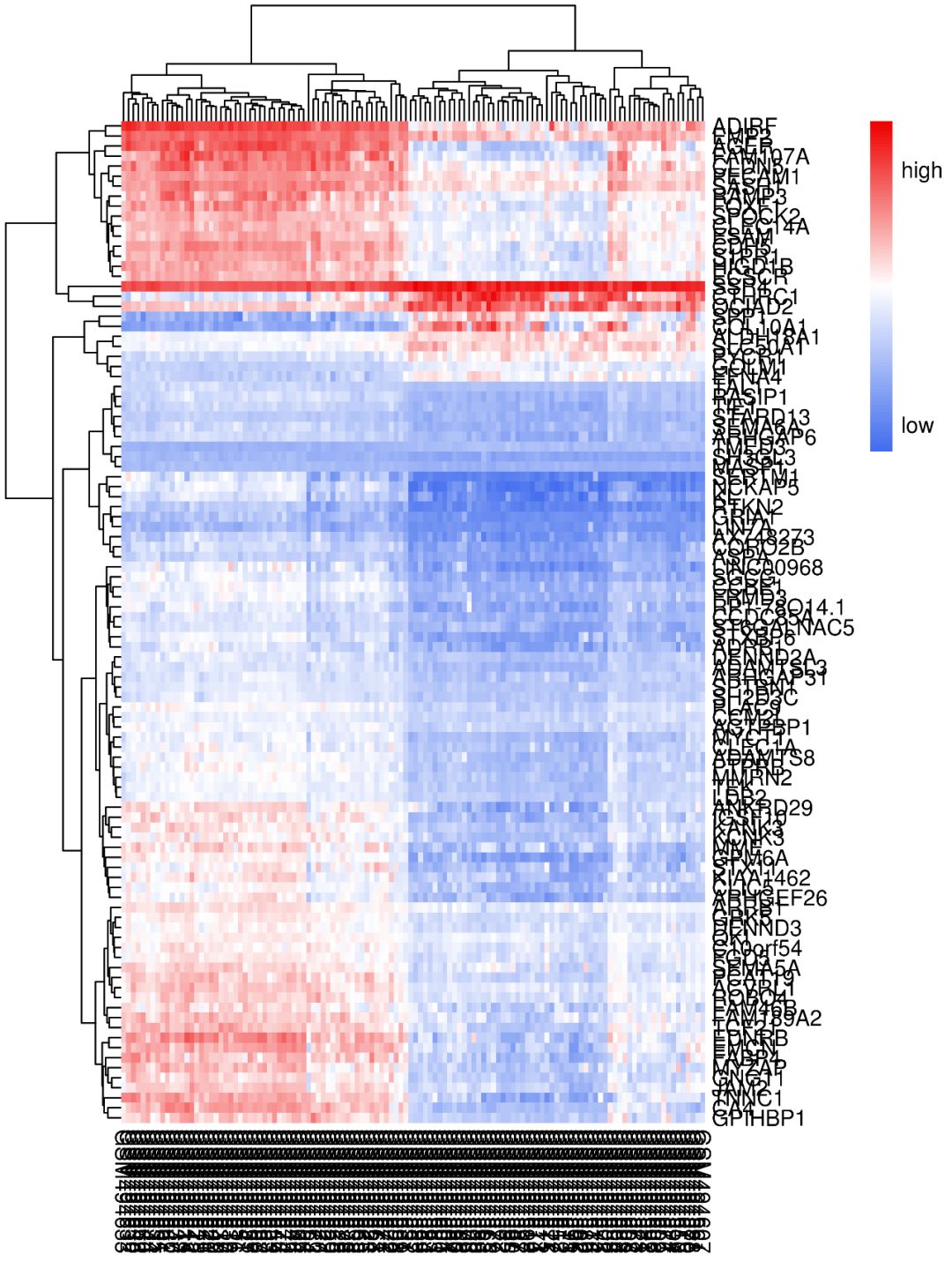
注释条
- annotation_colors:对标签的颜色进行修改
- annotation_legend:是否显示标签注释条
- annotation_row:数据框格式,用来定义热图所在行的注释条
- annotation_names_row:逻辑值,是否显示行标签名称
- annotation_col:数据框格式,用来定义热图所在列的注释条
- annotation_names_col:逻辑值,是否显示列标签名称
这里我们以列注释条为例。在注释条中,这里可以看到正常样本的大类中混有几个癌症样本,癌症大类中也混入了几个正常样本?你有没有想过是为什么呢?当然,你也可以选择不对列进行聚类,这样样本的顺序可以随你自己决定怎样放在一起。

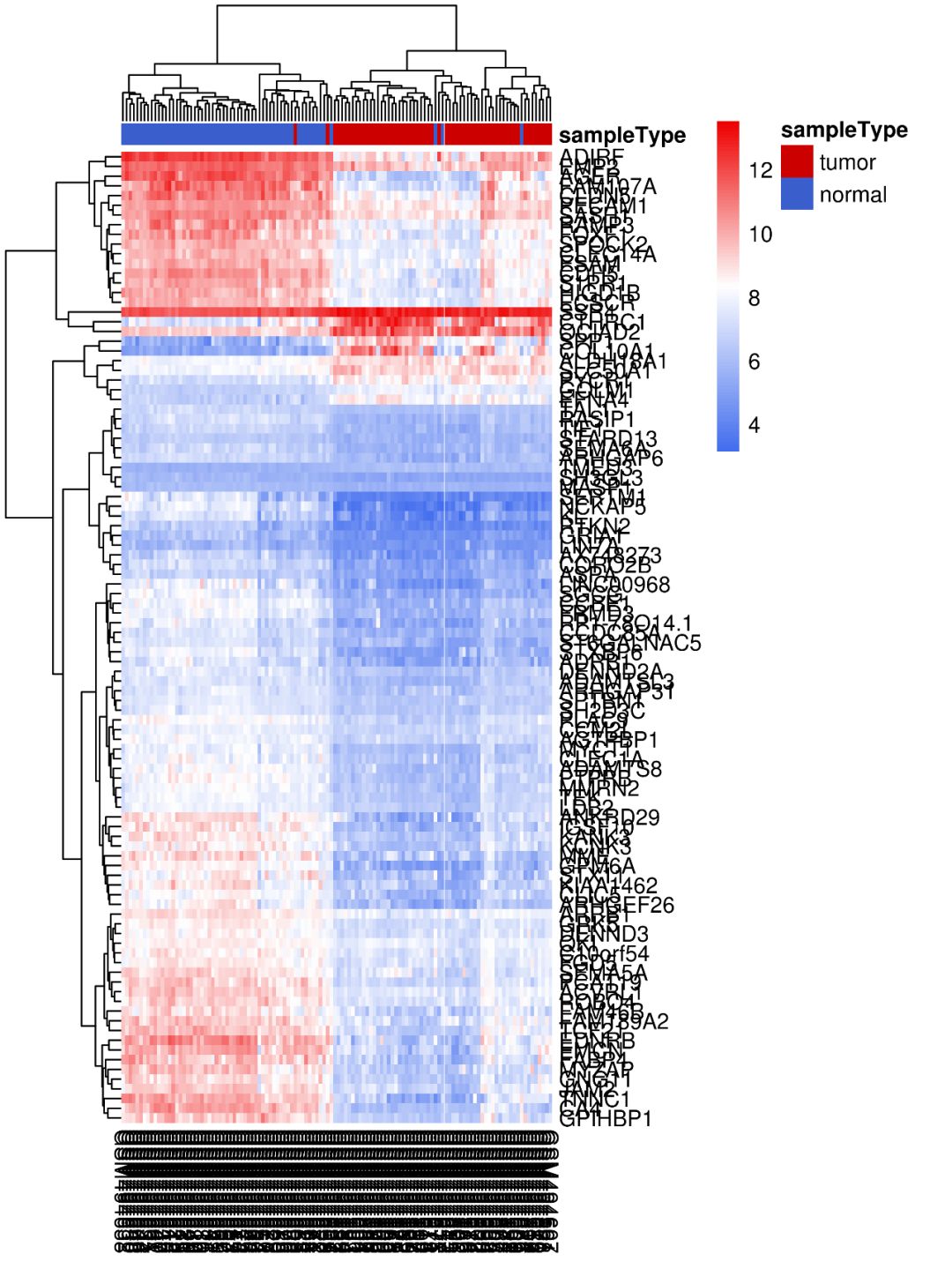
其他修改参数
- main:设置图的标题
- fontsize:是设置所有除主图以外的标签的大小
- number_color:字体的颜色
- show_rownames:是否显示行名
- fontsize_row:行名的字体大小
- labels_row:X轴坐标名设置
- show_colnames:是否显示列名
- fontsize_col:列名的字体大小
- labels_col:y轴坐标名设置
我们修改了字体大小,不显示样本名,以及设置图片标题,到此时,经过一系列参数更改,是不是已经比一开始使用默认参数好看多了呢?(可以增加一个对比图,GSE19804_tumor_vs_normal_heatmap6.png与GSE19804_tumor_vs_normal_heatmap1.png的 对比)。

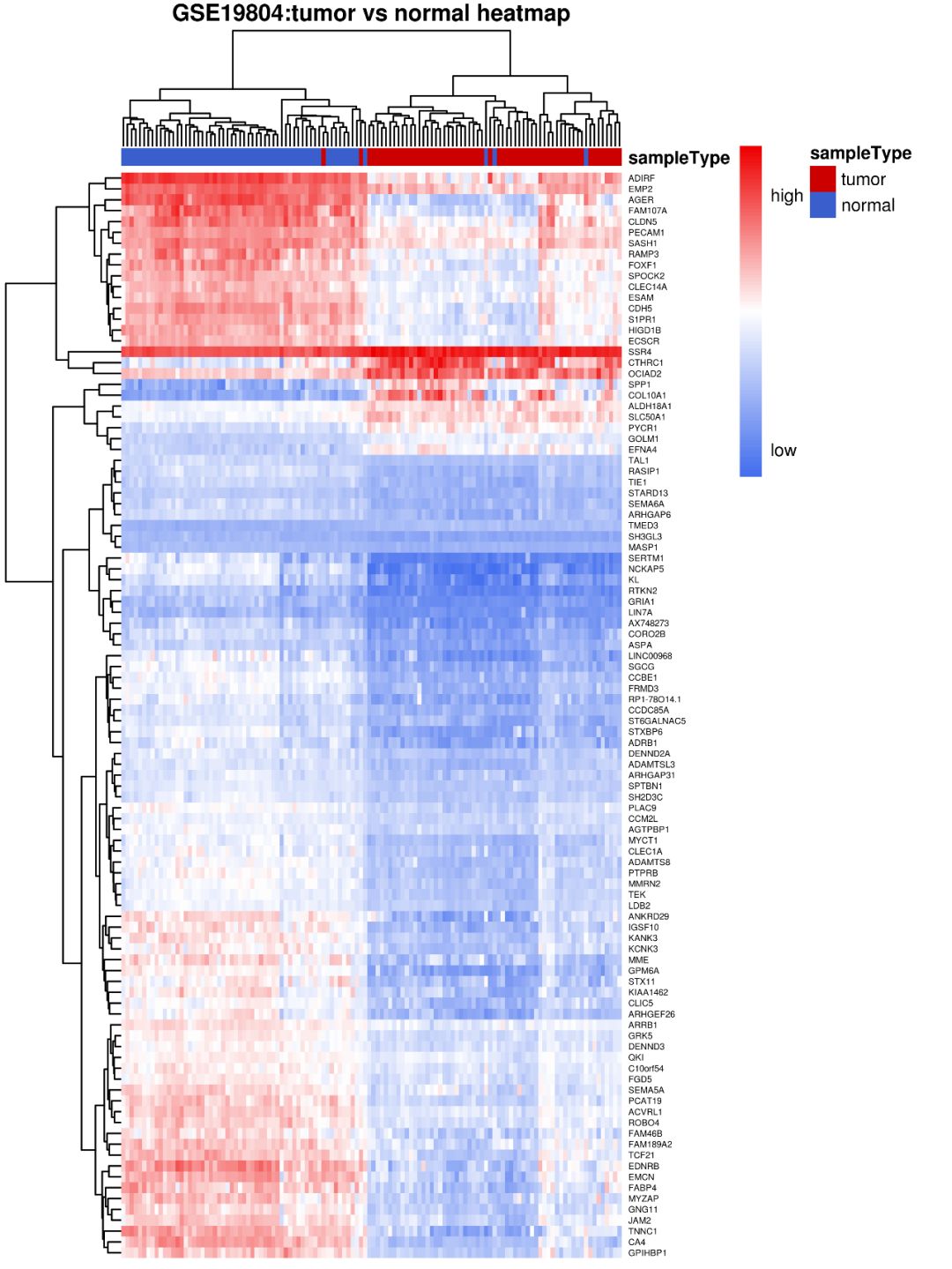
当然还有一些其他的用到不多的参数,留给读者自己去实验一下吧…
小格子参数设置
热图是由一个个的小四方格子组成的,每一个小格子代表一个基因在一个样本内的表达情况。
- fontsize_number:小格子中数字大小
- display_numbers:逻辑值,是否在小格子中显示数字
- number_format:小格子中数字显示形式,但仅有在display_numbers=T时才能使用
- na_col:设置小格子为缺失值时的颜色
- cellwidth:表示每个小格子的宽度
- cellheight:表示每个小格子的高度
输出文件参数设置
一般可以直接将画好的热图以png格式或者pdf格式进行写出。
- filename:输出图画的文件名
- width:输出图画的宽度
- height:输出图画的高度
不想看复杂的代码?想可视化一键绘图?那就用百迈客云的绘图工具啊。>>立即使用




 京公网安备 11011302003368号
京公网安备 11011302003368号