

 题目:Genome sequencing and comparativegenomics reveal the potential pathogenic mechanism of Cercospora sojina Hara onsoybean
题目:Genome sequencing and comparativegenomics reveal the potential pathogenic mechanism of Cercospora sojina Hara onsoybean
杂志名称:DNA RESEARCH
影响因子:5.415
合作单位:中国科学院等
研究背景
材料方法
从C.sojina侵染的大豆幼苗中分离出真菌单孢子。将分离的孢子在28℃下在马铃薯葡萄糖琼脂(PDA)培养基上生长。
2、基因组测序和组装:
二代(Illumina),270bp文库,90 x + 三代(PacBio)10kb文库,123x
3、基因组注释:
将所有预测的基因用数据库进行功能注释,数据库为:Swiss-Prot、NR、KEGG、COG、Pfam、HMMER、GO、PHIbase。
4、重复序列和全基因组DNA甲基化分析
使用TandemRepeats Finder(TrF)鉴定串联重复序列。使用三种软件Repeat Modeler、Repeat Protein Masker和Repeat Masker严格挖掘转座因子(TE)。
5、系统发育分析和共线性分析、碳水化合物活性成分的比较分析、次级代谢产物的提取和定量
6、转录组分析和定量RT-PCR:
7、推定效应子的功能研究
将推定的效应基因克隆到由35S启动子驱动的二元载体pMD-1中。通过电穿孔将构建好的载体转化到根癌土壤杆菌菌株EHA105中。使用含有指示效应物的EHA105菌株渗入4周龄本塞姆氏烟草叶无针注射器。GFP和Phytophothorasojae效应子Avr1b分别作为阴性对照和阳性对照。二十四小时后,用EHA105渗透植物携带pVX-BAX。
研究结果
1、C. sojina基因组的组装
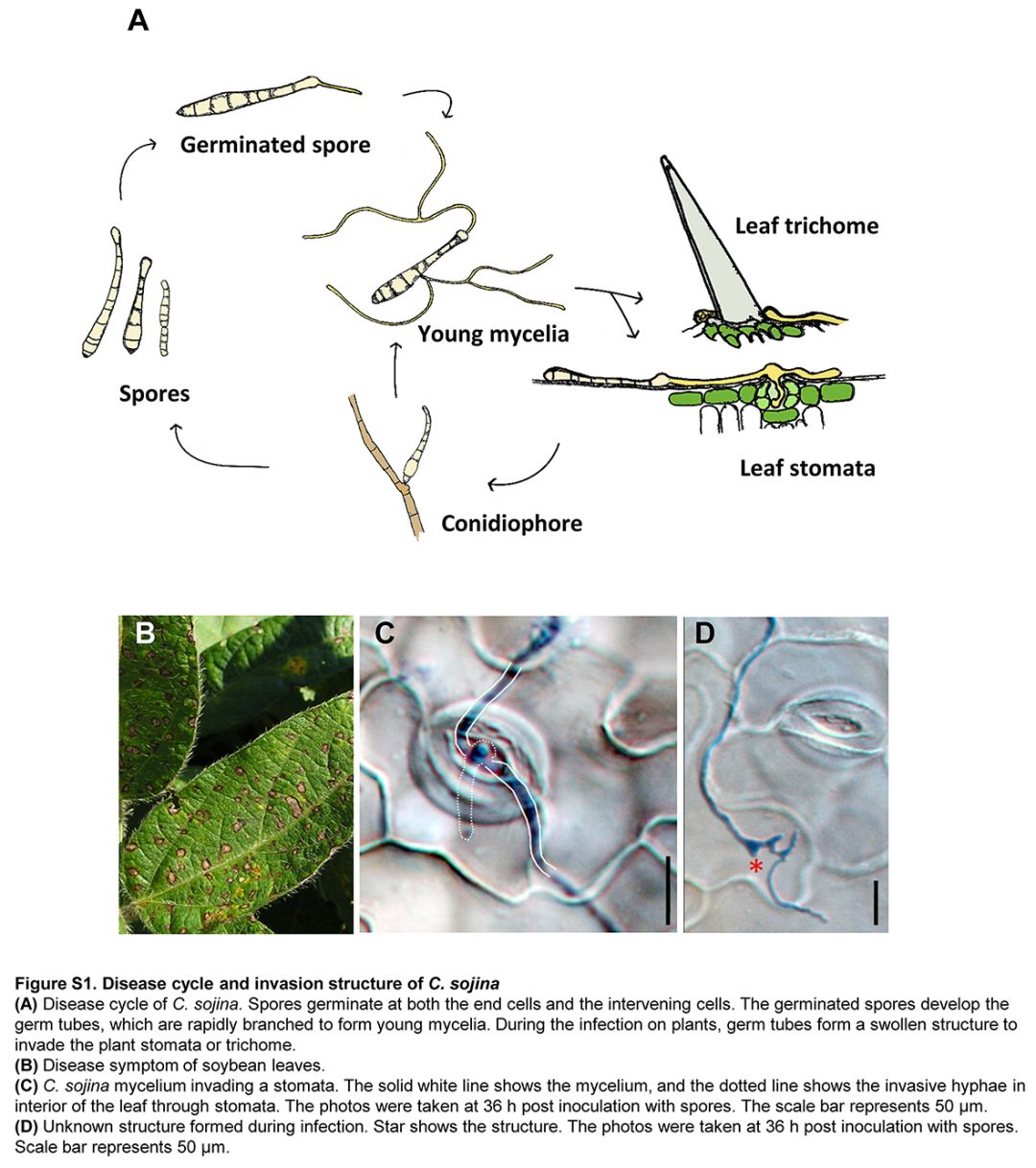
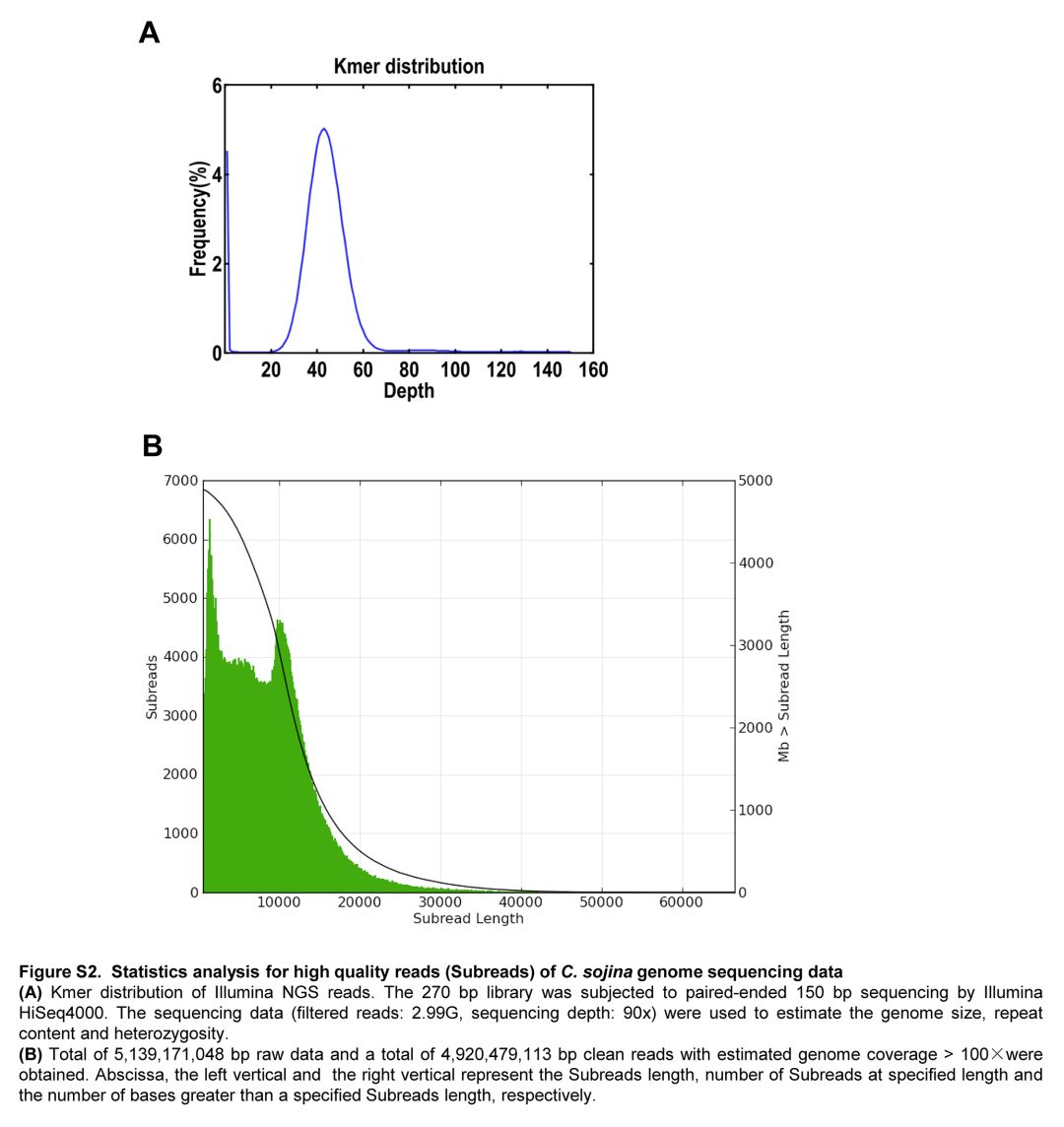
像大多数真菌一样,C. sojina表现出类似的感染周期,但它也表现出一些区别(图S1)。它不形成附着物,但分枝菌丝能通过开放气孔感染植物(图S1C和D)。与其他半营养真菌相比,FLS疾病发展相对较慢(图S1C)。Kmer峰图显示该菌为单核真菌。(图S2)
使用CANU从头组装测序数据(4,920,479,113bp clean reas),N50长度为1.59Mb,总组装大小约为40.84Mb(表1)。通过circos-plot显示了24个最大的支架(图1)。预测共11,655个蛋白质编码基因,其基因密度为每1 Mb约285个基因。在基因组中预测了277个tRNA和281个假基因。RNA-seq数据显示有8,474个推定的蛋白质编码基因。

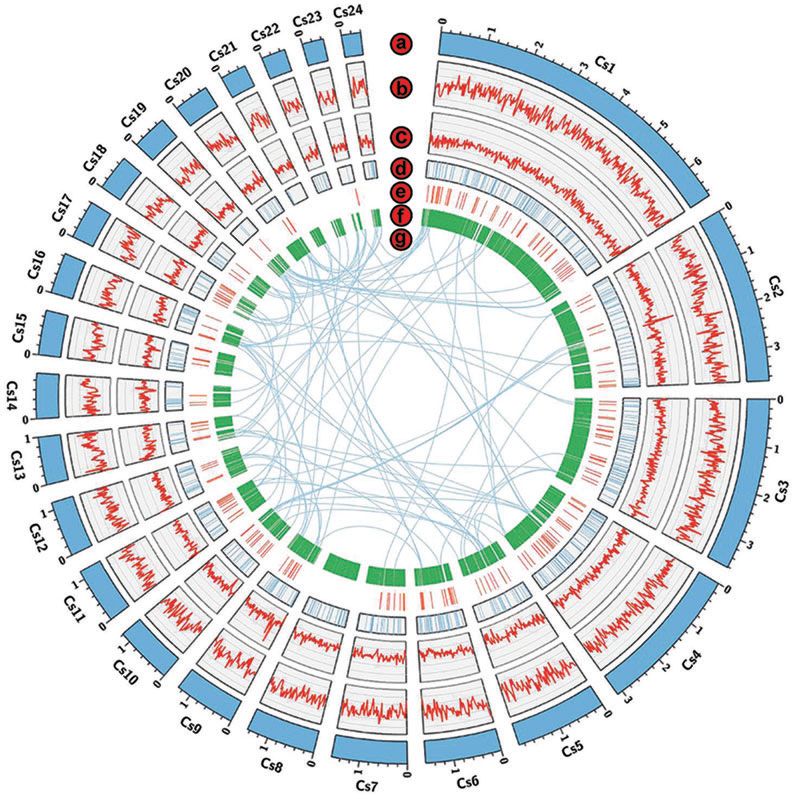
Figure1. Circos-plotof C. sojina.
2、重复序列和潜在的甲基化位点
重复DNA序列和TEs在真菌的进化,基因组结构和基因功能中起重要作用。在C.sojina基因组中鉴定了总共11,138,239bp(11M)重复序列,包括DNA转座子,LTR反转录转座子,串联重复序列和其他未分类的转座子(图2A)。重复序列占基因组的25.56%。大多数重复序列(96.36%)是TE,而串联重复序列仅占0.93%。DNA转座子和LTR反转录转座子分别占所有TE的28%和25%。
DNA甲基化涉及许多重要的细胞过程,例如基因组印记和基因转录调控。虽然已经在高等植物和动物中发现DNA甲基化多年,但最近在一些真菌中也有相关报道。使用SMRT能够检测到m6A和m4C甲基化。在C.sojina基因组中共鉴定了1,015,733m4C(4-甲基 -胞嘧啶)和17,409m6A(6-甲基 -腺苷)(图2B)。大多数分类的DNA甲基化是m4C,占98.3%,而m6A仅占1.7%。然而,与m6A相比,m4CDNA甲基化在重复元件的区域中出现低频率较低(图2C-E)。
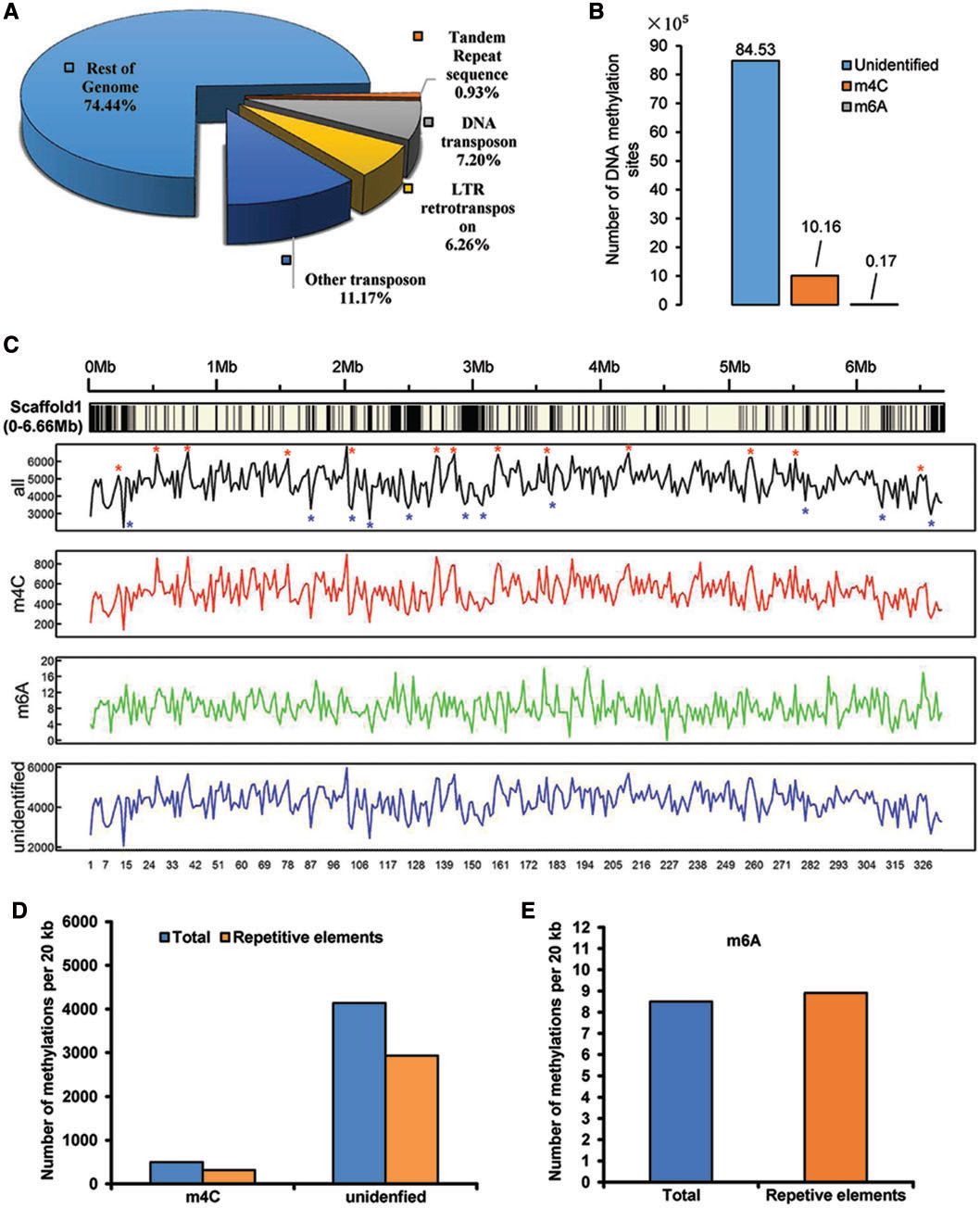
Figure2. Repeatelements and DNA methylation sites of C.sojina.
3、比较基因组分析
利用一组真菌的系统发育骨架基因分析了C. sojina和其他真菌物种的进化关系。系统发育分析表明C. sojina在进化上接近于尾孢菌(Cercospora zeae-maydis),一种可引起玉米叶斑病的植物病原体(图3A)。此外,C. sojina也接近其他三个病原体Pseudocercospora fijiensis,Sphaerulina musiva和Dothistroma septosporum(图3A)。
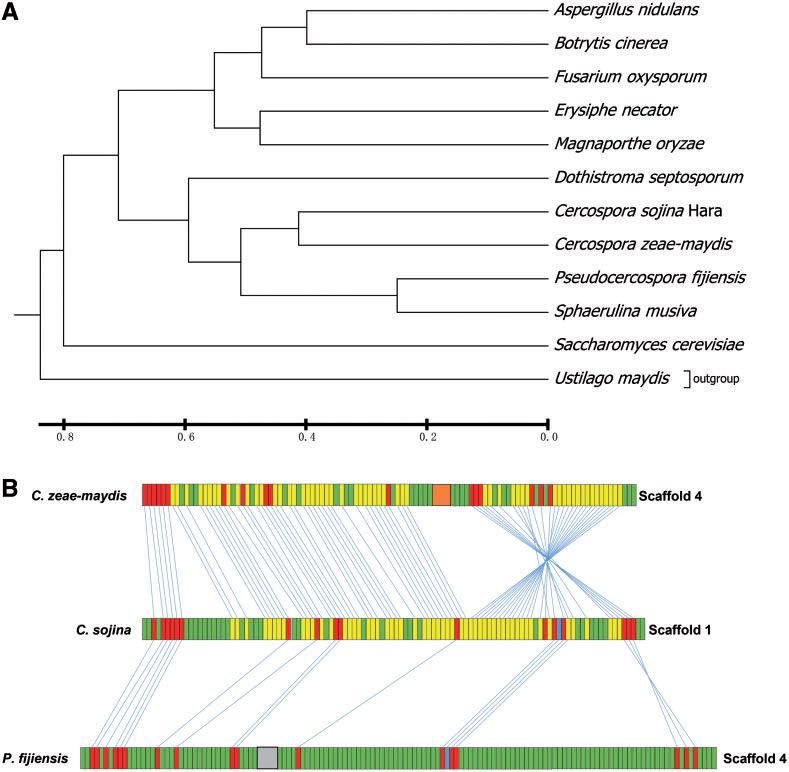
Figure 3.Phylogenetic and synteny analysis of C. sojina with other fungal species.
4.分泌组织和潜在的效应器
C. sojina的基因组包含11,655个蛋白质编码基因,覆盖约41%的基因组序列。最丰富的结构域是PF14295.4(n = 6),其介导蛋白质 – 蛋白质相互作用。其他常见结构域包括abhydrolase域(PF12697.5,n5),水解酶域(PF12146.6,n = 5)和PAN域(PF00024.24,n = 5)。
5.通过全基因组转录分析上调致病相关基因
由于C.sojina对大豆的感染进展非常缓慢,难以收集足够的样本来检查植物菌丝的基因表达。饥饿治疗可以模仿感染期间病原体的生理学。因此,使用生长24和48小时的菌丝体,并通过RNA测序进行转录组分析;分别在饥饿处理后24小时和48小时鉴定出3,227和3,223个差异表达的基因(DEG)。四类DEG引起了我们的注意,这些基因注释为参与PHI,分泌蛋白组,推定的碳水化合物活性酶(CAZymes)和次级代谢过程。
6.基因簇用于次级代谢物
尾孢菌(Cercosporasojina)基因组编码16个非核糖体肽合成酶(NRPS),20 个PKS,18个脂肪酸合成酶,3个萜烯合酶,2 个geranylgeranyl二磷酸合成酶和1种萜类化合物环化酶。这些酶参与次级代谢产物的合成,包括霉菌毒素,色素和生物碱。在C. sojina基因组中鉴定了一个具有8个尾孢菌素生物合成基因的类似基因簇。(图4A)。这八个基因显示高氨基酸序列与烟草(C. nicotianae)尾孢菌素生物合成基因的相似性在相同的串联顺序中(图4A)。此外,我们观察到感染期间8种基因的转录增加(图4B)。
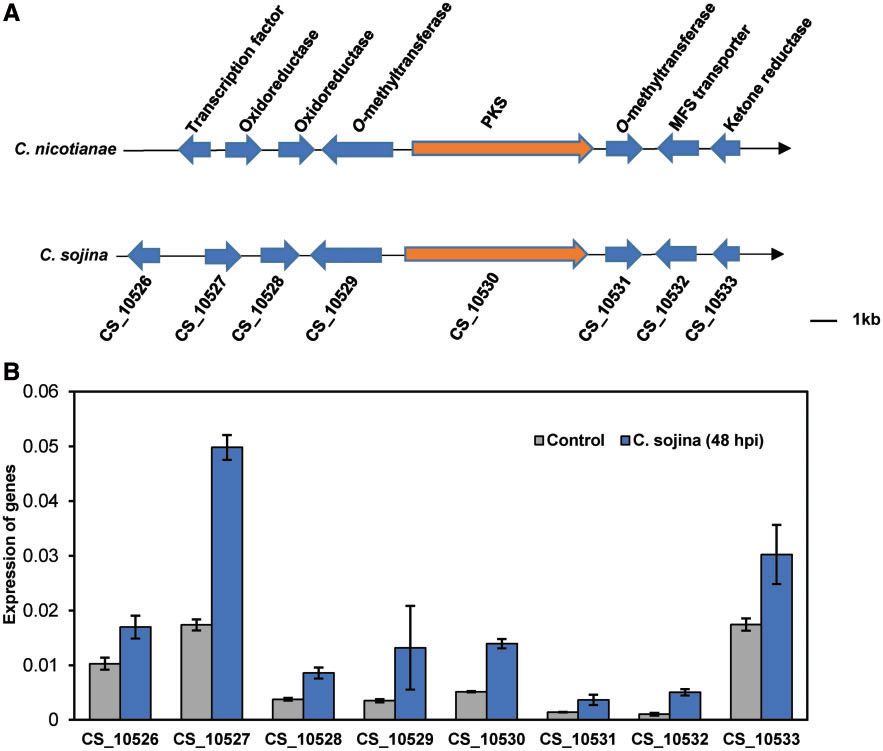
Figure 4. Putative gene clusters forcercosporin biosynthesis in C. sojina.
色素是成功入侵病原体的另一组次要代谢物。通常,病原体产生的色素能够在感染期间保护病原体免受宿主氧化应激。C.sojina基因组编码多个推定的PKS,其负责色素产生(图5A)。C.sojina还可产生一些灰色色素,并且通过饥饿和cAMP处理显著诱导色素(图5B),表明色素可能与病原体毒力有关。因此,进一步分离和部分纯化了色素。得到灰色,浅黄色和深灰色三种色素,深灰色是最丰富的色素。
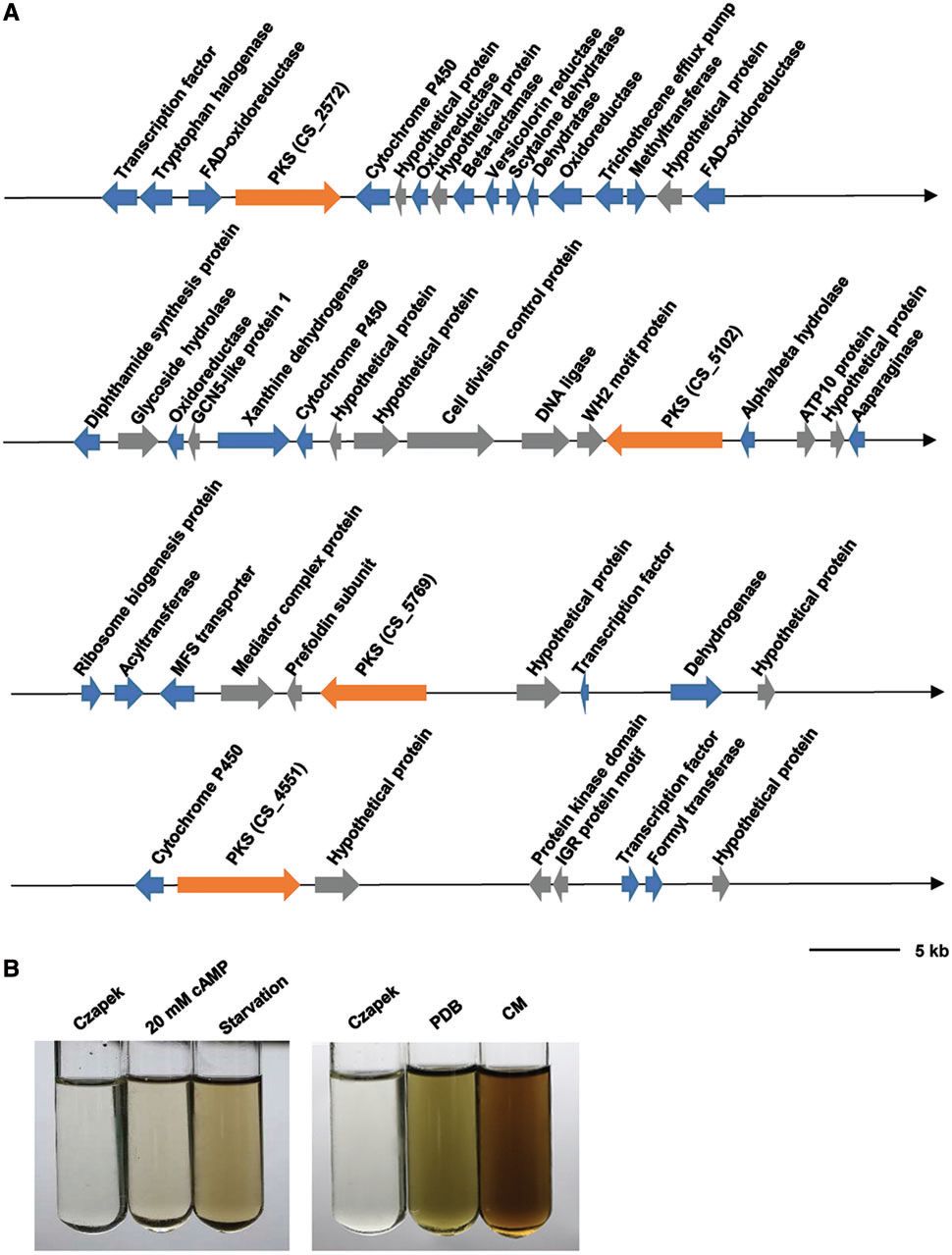
Figure 5. PutativePKS gene clusters for pigment production in C.sojina.
7.碳水化合物活性酶
成熟的植物致病真菌可以分解并利用CAZymes的植物细胞壁多糖。尾孢菌(Cercospora sojina)含有596个预测的CAZymes。与其他真菌相比,C.sojina具有更大的潜在碳水化合物酯酶组,其可催化取代糖的N-去酰化。在C. sojina基因组中,有大约23.5%的潜在分泌蛋白被预测为CAZyme,证明C.sojina可能在入侵期间使用大量CAZymes来消化宿主细胞壁。有趣的是,其中一个CAZymes家族,即糖苷水解酶GH109家族,是高度富集的(图6)。
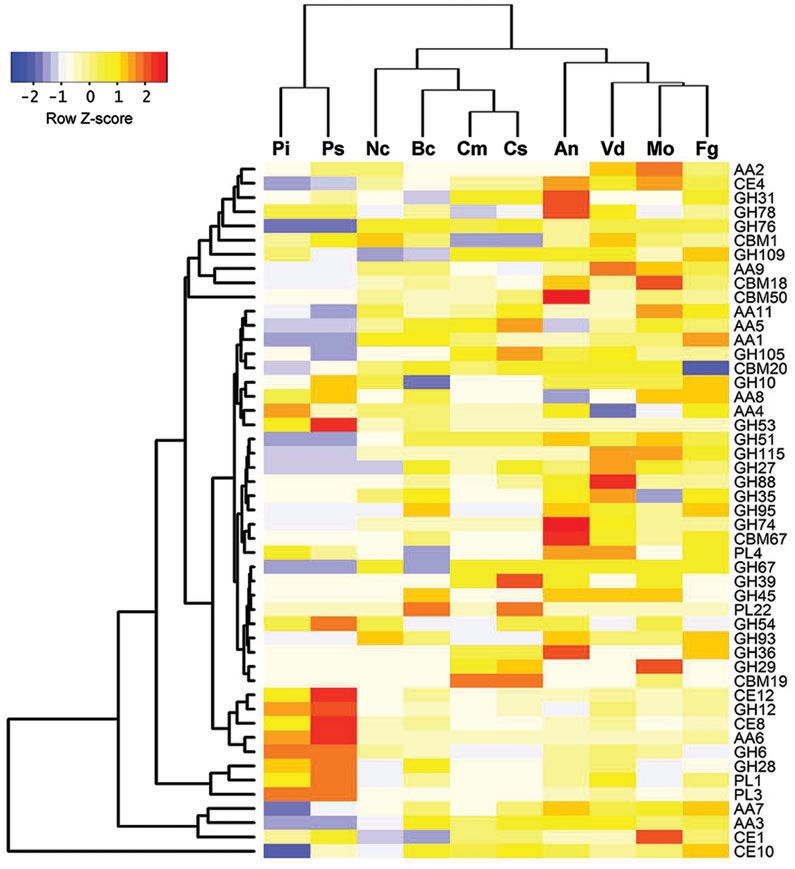
Figure 6. Comparison of carbohydrateenzymes between C. sojina and nine other fungal species.
8.推定效应子的功能分析
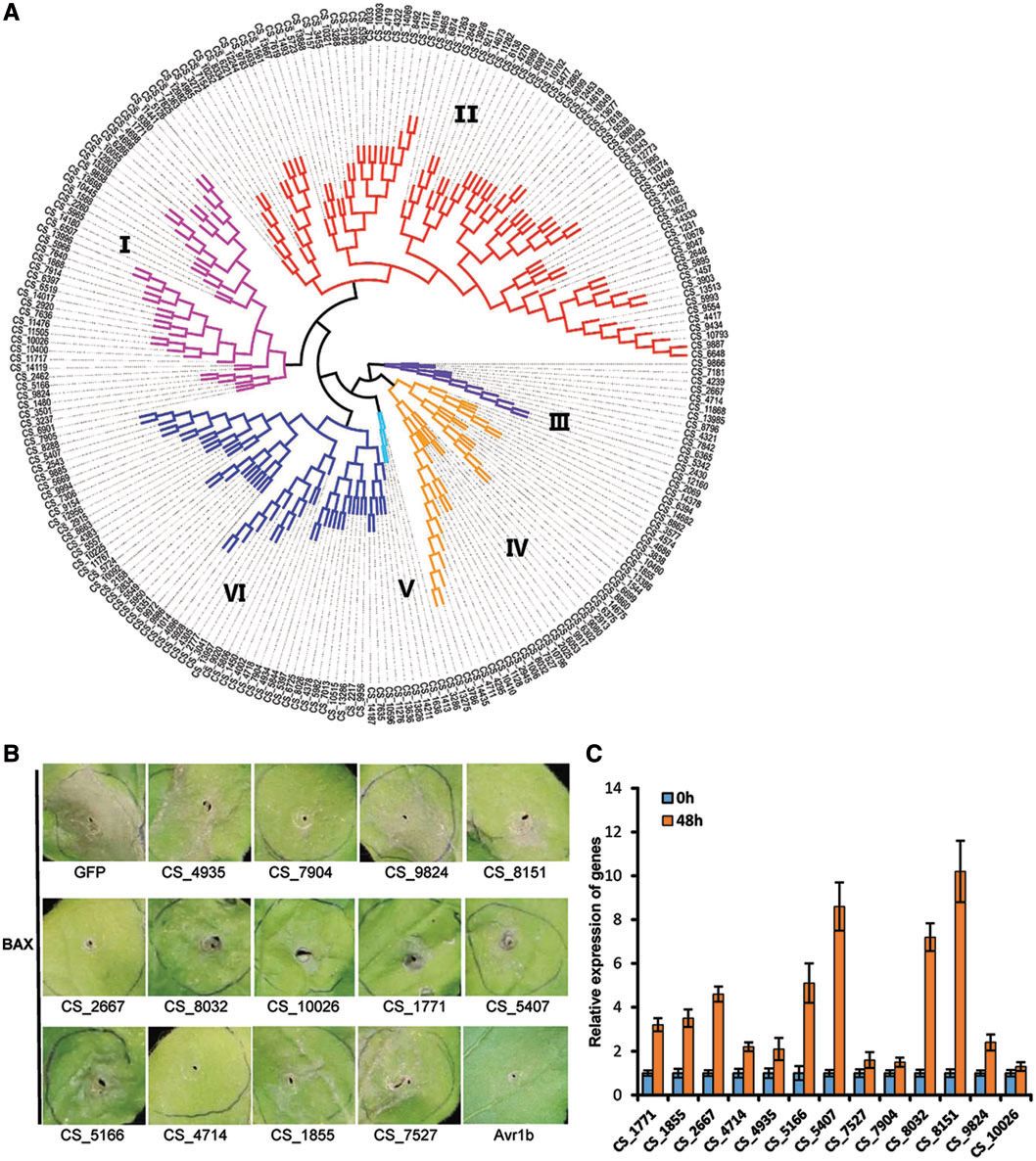
效应器是由细菌,卵菌或真菌分泌的低分子量蛋白质,其损害宿主免疫防御并适应特定环境。在玉米病原体U.maydis中,观察到分泌的蛋白质编码基因簇中的大多数基因在感染组织中同时被诱导。233个推定效应子的系统发育分析显示,21个预测的效应子被分组成在C.sojina中含有2或3个高序列相似性基因的簇(图7A)。假定的效应子群集也表明局部重复可能参与了C.sojina中效应子的扩增。另外,40个推定的效应器可以通过PHI数据库注释,并且大多数注释的效应子与真菌发病机理有关。
因此,我们尝试研究效应器功能。促凋亡小鼠蛋白BAX诱导的本氏烟草程序性细胞死亡(PCD)在生理上可能与病原体引起的防御相关的过敏反应相似,提供了有价值的筛查可以抑制与防御相关的PCD的效应物的方法。我们随机选择50个效应子并在本塞姆氏烟草中瞬时表达它们以筛选可以抑制BAX触发的PCD(BT-PCD)的潜在效应物。结果表明,约1/4选择的效应子强烈抑制BT-PCD(图7B)。此外,qRT-PCR结果显示,大多数这些推定的效应子在C.sojina感染后48小时被转录诱导(图7C),这意味着它们可能有助于大豆灰斑病的早期感染。
结论
本研究通过SMRT测序技术组装了C. sojina的完整基因组序列。这种测序技术不仅有助于我们找到重复序列,还有助于发现真菌中的DNA甲基化。通过基因组组装和注释,预测特定的CAZymes,次级代谢产物和效应子可以帮助C. sojina成功的适应大豆。同时,本研究也为这一重要的大豆灰斑病未来防治及发展奠定了基础。
Luo X ,Cao J , Huang J , et al. Genome sequencing and comparative genomics reveal thepotential pathogenic mechanism of Cercospora sojina Hara on soybean[J]. DnaResearch, 2018, 25(1):25-37.
如果您的项目遇到问题,欢迎点击下方按钮咨询我们,我们将免费为您设计文章方案





 京公网安备 11011302003368号
京公网安备 11011302003368号