
从1977年第一代测序技术的问世,发展至今已有四十余年,这几十年间,测序技术的发展突飞猛进,从一代到二代再到三代,每一步突破都是测序行业一次质的飞跃。三代测序以PacBio公司的SMRT测序技术为代表,测序过程无需PCR扩增,再加上三代测序长读长的优势,无需组装,可以直接获得从5’端到3’端高质量的全长转录组信息,可准确鉴定基因的可变剪接、APA(可选择性多聚腺苷酸化)、融合基因、基因家族和非编码RNA等信息。百迈客2016年率先引进三代测序平台PacBio RSⅡ和PacBio Sequel,至今已积累了丰富的项目经验,无论实验能力还是信息分析能力都达到行业较高水平。一定不是小编吹嘘,咱们用事(案)实(例)说话。
案例一:
Identification and analysis of glutathione S-transferase gene family in sweet potato reveal divergent GST-mediated networks in aboveground and underground tissues in response to abiotic stresses通过鉴定和分析谷胱甘肽S转移酶基因家族揭示甘薯地上和地下组织响应非生物胁迫中的GST介导的网络调控机制
发表杂志:BMC Plant Biology
影响因子:3.964
发表时间:2017
实验材料:甘薯
测序方法:2+3联合测序
文章概述:基因重复是物种进化研究的重要内容之一。基因重复可以导致基因在编码蛋白以及表达量上的多样性,并最终导致明显的功能分化。某个基因家族可能是由同一个祖先(全基因组或单个基因)通过基因重复分化而来。谷胱甘肽S-转移酶(GSTs)是一类基因家族,其广泛真核生物和原核生物中。GST基因可以参与到生物体响应非生物胁迫中(如臭氧、过氧化氢、植物激素、重金属、热激、干旱等)。甘薯是六倍体物种,其复杂的遗传背景被认为是多个物种的全基因组重复形成的。本文通过2+3转录组测序技术进行基因家族分析。发现GST基因广泛参与甘薯的非生物胁迫相应中,且层次聚类分析表明GST基因的在甘薯地上和地下组织中响应非生物胁迫的表达模式明显不同。
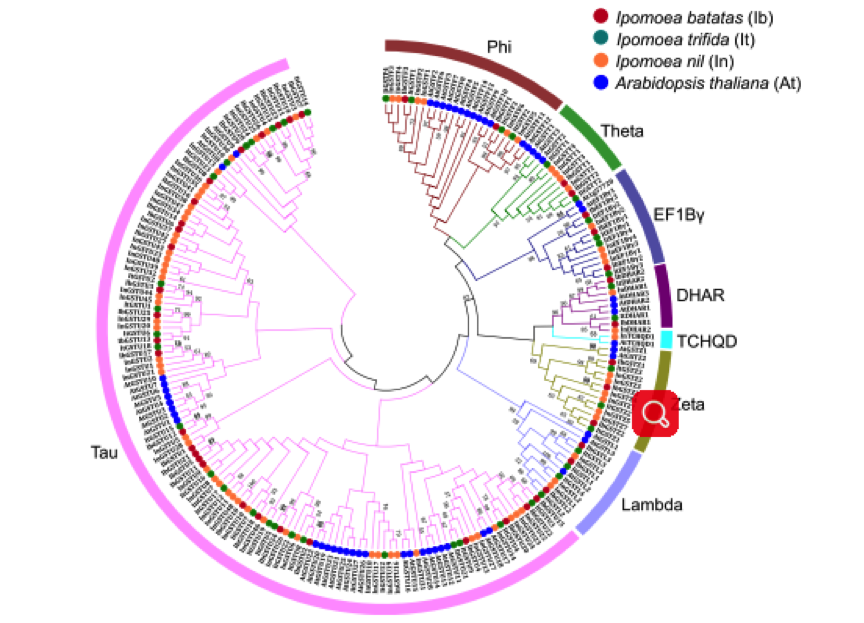
图1. 甘薯、野生甘薯、拟南芥、牵牛花GST蛋白系统进化树
案例二
SMRT sequencing of full-length transcriptome of flea beetle Agasicles hygrophila (Selman and Vogt)
莲草直胸跳甲全长转录组分析
发表杂志:Scientific reports
影响因子:4.259
发表时间:2018年1月
实验材料:莲草直胸跳甲
测序方法:2+3联合测序
文章概述:空心莲子草Alternanthera philoxeroides (Mart.) (Amaranthaceae)是多年生杂草,原产于南美洲,20世纪30年代传入我国。在中国空心莲子草被认为是一种重要入侵物种,并且严重影响了农业生产和生态环境。莲草直胸跳甲Agasicles hygrophila是空心莲子草的专性天敌,作为生物防治手段而被引入。目前,莲草直胸跳甲的基因组和转录组信息尚未被研究,因此在本研究中作者对其进行了全长转录组测序,获得较完整的转录本集合,预测了145个可变剪接事件;27,318条简单重复序列,经TransDecoder鉴定获得24,040个ORF,其中有16,205个完整的ORF;预测得到4,198 个lncRNA,为进一步研究莲草直胸跳甲与宿主植物和生态系统之间相互作用的分子机制提供了很好的遗传信息基础。
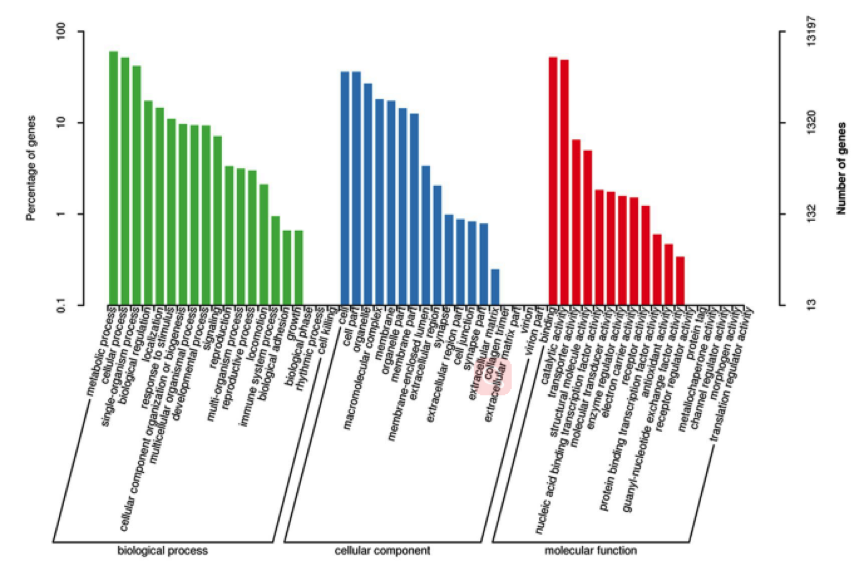
图2. 莲草直胸跳甲转录本的GO功能注释
案例三
Generation and comparative analysis of full-length transcriptomes in sweetpotato and its putative wild ancestor I. trifida甘薯及野生型甘薯的全长转录组比较分析
发表杂志:暂无开源期刊,冷泉港实验室在线发表
发表时间:2017年3月
实验材料:甘薯和野生甘薯
测序方法:2+3联合测序
文章概述:甘薯是许多发展中国家最重要的作物之一,也是重要的能量来源。然而,甘薯是同源六倍体植物,基因组大小约3-4G,目前还没有高质量的参考基因组。此外,甘薯基因组的杂合度高,目前还没有关于甘薯正向遗传学研究的报道。目前,没有获得大规模的全长cDNA序列阻碍了甘薯功能基因组学和分子育种研究的进展。长期以来,人们一直认为甘薯最有可能由二倍体祖先野生甘薯(I. trifida)进化而来,但是没有确凿的证据。该研究通过2+3联合测序的方法获得甘薯和野生甘薯的全长转录本,揭示二者的进化关系。甘薯和野生甘薯大多数的转录本都是同源的,并且确定了1269个假定的甘薯和野生甘薯之间的同源基因对,通过Ka/Ks比值分析,只有56个基因对的Ka/Ks比值大于1,表明在甘薯的进化或驯化过程中,大多数基因都是受到纯化选择的。
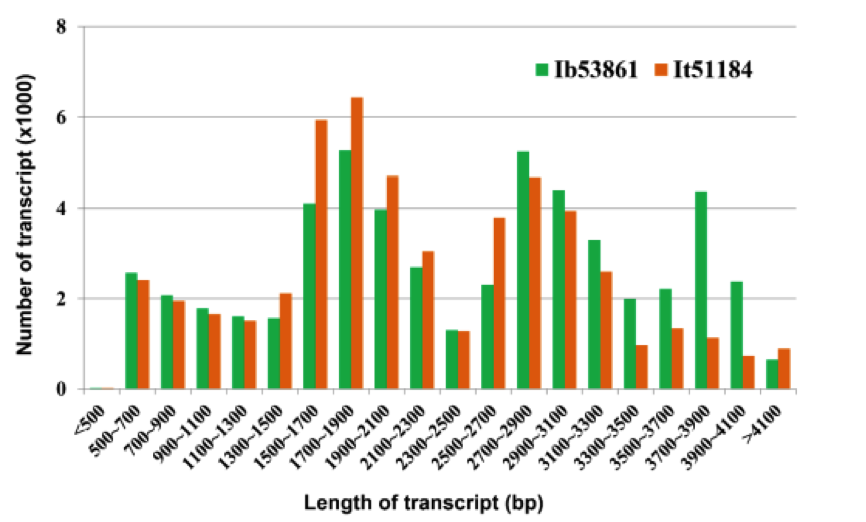
图3. 在甘薯(Ib53861)和野生甘薯(It51184)中的53,861个非冗余转录物的数量和长度分布。
案例四
A transcriptome atlas of rabbit revealed by PacBio single-molecule long-read sequencing通过PacBio单分子长读长测序技术揭示家兔转录本图谱
发表杂志:Scientific reports
影响因子:4.25
发表时间:2017
实验材料:兔子
测序方法:2+3联合测序
文章概述:兔子(Oryctolagus cuniculus),是重要的哺乳动物,由于其与人类系统发育关系密切,并且具有生命周期短、性格温顺等特点,因此在生物医学研究中将兔子作为模式动物。兔子的基因组大小为2.66Gb,从Ensembl数据库预测到22,668个基因位点包括了24,946个转录本,但是其中的大部分只是预测,不同的isoforms和非翻译区还缺少可靠的注释,对复杂的转录本评估不准确,通过高通量测序的方法可以较好的研究转录本、isoforms和基因表达水平,但是由于二代测序的短读长很难获得转录本全长,研究可变剪接和APA事件就成为问题,本研究利用PacBio三代测序平台,共鉴定到多达24,797个AS事件和11,184个APA事件,提供了一整套全面的转录本参考数据集,用于绘制兔的转录本图谱。对于改进兔基因组的注释有较大帮助。
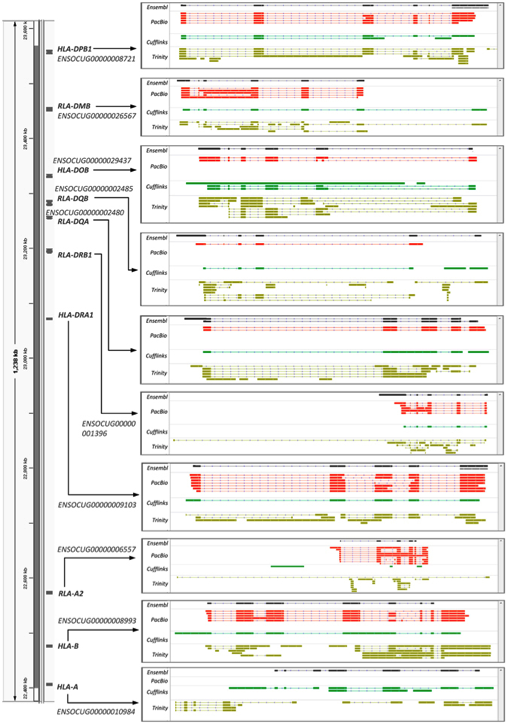
Figure 4.基因通过PacBio所测转录本和组装得到的转录本还原10个MHC基因。染色体定位、命名和每个基因的Ensembl编号(在左侧)
我们都知道好的测序数据要配上专业的分析团队才能让故事叙述的更加完美,百迈客研发团队于2018年5月在之前的分析基础上新增了一些分析条目,让您的数据抒写更华丽的篇章(如下表)。
百迈客新增分析服务表
| 标准分析(有参为例) | 新增分析内容 | 新增分析图 |
|---|---|---|
| 1.测序Reads质量质控(QV); | 1.分析转录本和基因的关系(无参全长) | 1.染色体 |
| 2.全长转录本评估; | 2.全长序列的完整性评估 | 2.基因组中基因的密度热图 |
| 3.Isoform序列聚类; | 3.转录因子分析 | 3.三代测序结果中测到的基因的密度热图 |
| 4.转录本去冗余; | 4.可变剪切分析结果可视化 | 4.基因组中转录本的密度热图 |
| 5.与参考基因组进行比对 | 5.等位基因分析(有参全长) | 5.三代测序结果中测到的转录本的密度热图 |
| 6.基因结构优化; | 6.SNP分析(无参) | 6.所有差异转录本在染色体上的分布密度直方图 |
| 7.新转录本预测; | 7.APA上下游碱基分布及motif分析 | 7.LncRNA在染色体上的分布密度直方图 |
| 8.可变剪接分析; | 8.差异可变剪切 | 8.融合转录本连线图 |
| 9.转录本功能注释; | 9.circos图展示 | |
| 10.融合转录本分析; | ||
| 11.基因家族分析; | ||
| 12.lncRNA预测; | ||
| 13.lncRNA靶基因预测; | ||
| 14.SSR分析; | ||
| 15.CDS预测; | ||
| 16.可变多聚腺苷酸化分析(APA); | ||
| 17.不同样品可变剪接差异对比(两个或以上样品); | ||
| 18.不同样品融合转录本差异对比(两个或以上样品); | ||
| 19.不同样品可变多聚腺苷酸化(APA)差异对比(两个或以上样品) |
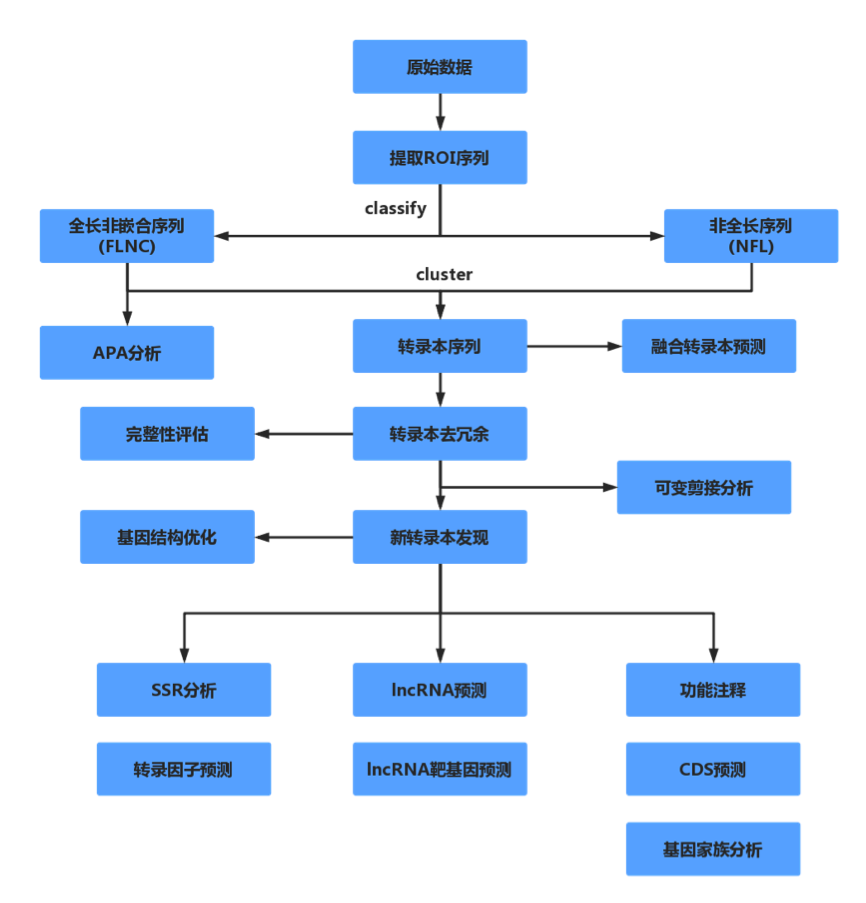
百迈客全长转录组分析流程图:
如果您想与我们的生信工程师沟通思路,请点击下面按钮,我们将免费为您设计方案




 京公网安备 11011302003368号
京公网安备 11011302003368号