


捕获Hi-C测序鉴定33个乳腺癌风险基因座潜在的110个靶基因
1.研究背景
目前GWAS研究已经鉴定了约100个乳腺癌易感基因座。绝大多数乳腺癌风险SNP位于非编码区,可能影响基因转录调控;甚至有许多风险SNP位于“基因沙漠”中,即距离最近的蛋白编码基因数百Kb远。鉴定这些风险位点作用的靶基因有助于深入理解这些GWAS风险位点的作用机制。鉴定乳腺癌风险位点功能的系统性方法,主要包括大基因组区域的精细定位、SNP基因型与邻近基因表达之间的相关分析(eQTL,基因表达数量性状定位)和染色质构象捕获技术(Hi-C、CHIA-PET等)鉴定调控区域的靶基因。
2.研究方法
利用Capture Hi-C(CHi-C)技术在6种细胞系中对68个乳腺癌风险基因座进行高通量测序,即首先构建Hi-C文库,针对该68个loci(目标区域共计14.7Mb)设计RNA捕获探针(Agilent),杂交捕获目标片段,最后上机测序,off-target(目标区域以外的) di-tags(插入片段两端的Reads)定义为2端都未比对到风险基因座。
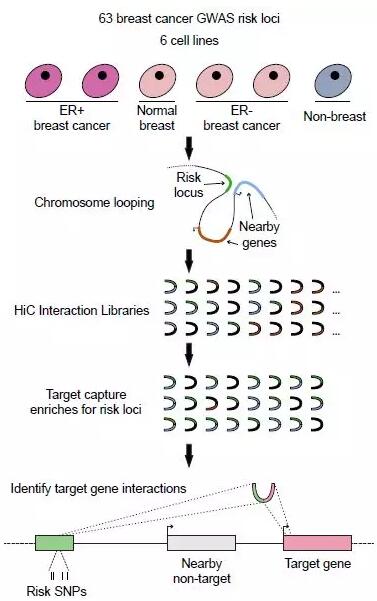
3.技术路线
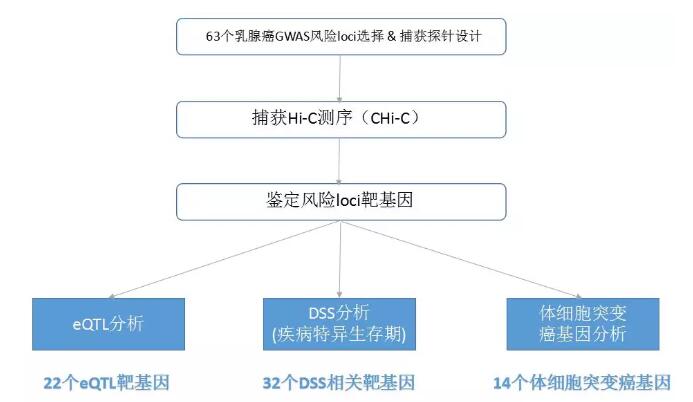
4.研究结果
1、63个风险基因座的互作peak(loop)分布
在2种ER +乳腺癌细胞系(T-47D,ZR-75-1)、2种ER-乳腺癌细胞系(BT-20,MDAMB-231)、1种“正常”乳腺上皮细胞系 (Bre80-Q-TERT/Bre80)和1种非乳腺类淋巴母细胞系(GM06990)中进行了CHi-C测序。风险基因座定义为包含至少1个风险SNP位点在内,且包含与该SNP相关(r2>0.2)的所有SNP的一个连续区域。每个细胞系的互作peak数目在0-1744的范围内,有12个基因座没有在以上任何一种细胞系中鉴定到互作peak。剩余的51个loci中46个是与所有乳腺癌或者ER+乳腺癌风险相关的。
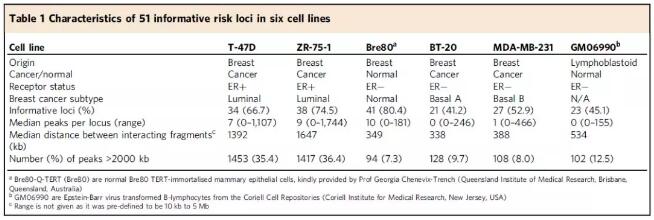
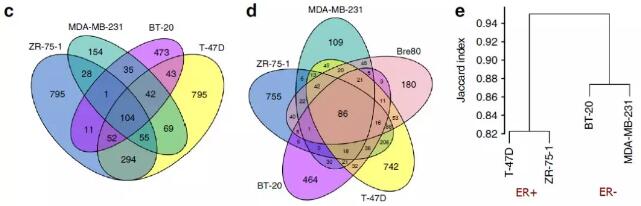
作者发现ER+乳腺癌细胞系中互作peak数目显著高于ER-乳腺癌细胞系,且ER+乳腺癌细胞系中存在很大一部分距离大于2Mb的互作片段。分析样本间互作peak的Jaccard相似性,发现ER+和ER-各自组内具有更高的相似性。
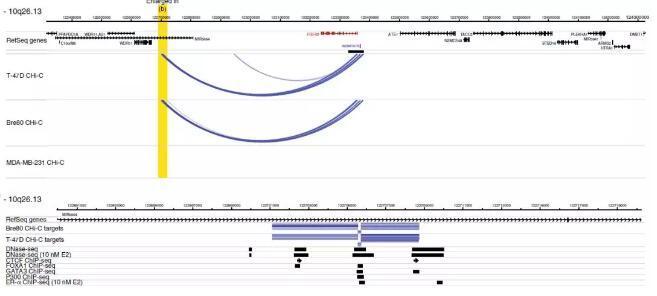
互作peak示例:下图为10q26.13基因座的互作peak(loop),在ER+乳腺癌细胞系和正常乳腺上皮细胞系中鉴定到了互作peak,但是ER-乳腺癌细胞系中则未检测到。互作靶基因的转录起始位点(TSS)位于捕获区域内,并且和相距几百Kb远的特异的HindIII酶切片段形成环(互作Peak)。在T-47D细胞系中,这些远距离片段与DNase I超敏位点、CTCF、FOXA1、GATA3转录因子结合位点以及雌激素受体ERα结合位点共定位。
2、定义潜在的靶基因
作者将靶基因定义为至少两个细胞系中,定位到捕获区域内或捕获区域的顺式作用区间(≤5 Mb)的基因,且其转录起始位点/TSS定位在相互作用片段上。以此为准能够为51个基因座中的33个(64.7%)鉴定到110个推定的靶基因,其中94个为蛋白编码基因,16个非编码RNA。作者比较了GWAS风险SNP位点的临近基因以及本研究中鉴定到的靶基因,发现其中24个基因座的邻近基因是唯一的CHi-C靶基因(N = 9)或几个CHi-C靶基因之一(N = 15)。
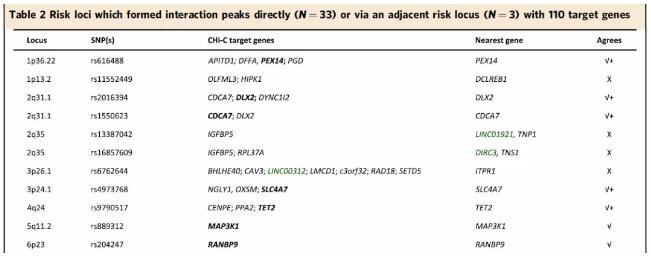
注:√表示邻近基因与CHi-C靶基因一致,且有且仅有一个靶基因;√+表示该基因座邻近基因为CHi-C靶基因之一;X 表示邻近基因不是CHi-C靶基因。
3、CHi-C靶基因和eQTL分析
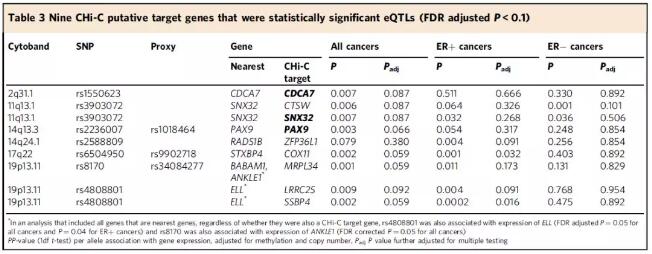
为了评估推定的靶基因在乳腺癌病因中具有因果作用的可能性,作者首先使用风险SNP(或与风险SNP连锁不平衡的位点,r2> 0.8)和TCGA数据库中的RNA-Seq数据(N=547)进行eQTL分析( TCGA),并对匹配的DNA甲基化数据和体细胞拷贝数变异进行了校正,根据ER状态进行分层分析。共计鉴定到了22个显著SNP-基因组合(t检验p<0.05),其中9个经过多重检验FDR校正后仍然显著。其中,3个基因为邻近基因。
4、CHi-C靶基因和疾病特异性生存期(DSS)分析
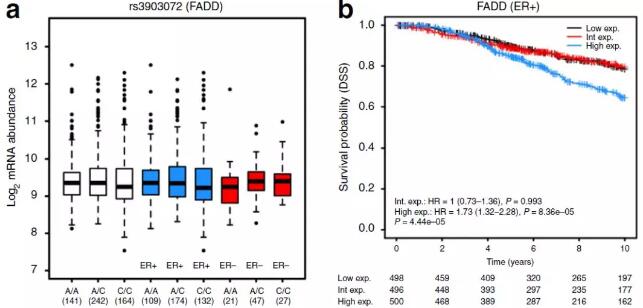
作者所纳入的风险SNP在此之前只有一个位点(16q12.1-rs3803662)报道了与疾病预后相关。本研究中作者直接分析了代谢型乳腺癌队列中潜在的靶基因的表达水平与的患者预后之间的关联(分别对ER +和ER-亚组进行生存分析)。其中32个靶基因(33%)与ER +乳腺癌患者的DSS相关,6个DSS相关的靶基因为eQTL靶基因。
5、CHi-C靶基因和体细胞突变癌基因分析
CHi-C推定的靶基因与560个乳腺癌的全基因组测序分析得到的727个癌基因列表进行比较,CHi-C靶基因高度富集这些癌症基因(n=14),包括证据充分的癌基因(CCND1、 CDKN2A、CDKN2B、MYC、MAP3K1、 ESR1和FGFR2)以及表征不明的癌基因(TET2、 KLF4、 MLLT10、FADD、TBX3、PAX9 和 ZFP36L1)。
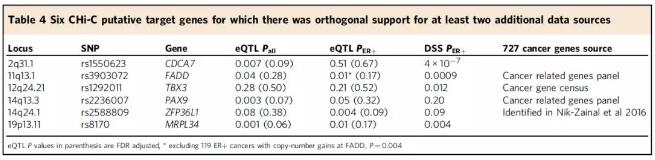
综合分析体细胞突变数据与eQTL和DSS三种指标,6个基因座至少有2个指标支持,见下表。其中4个基因CDCA7、FADD、ZFP36L1和MRPL34的表达水平同时与SNP基因型以及DSS相关。
怎么样,Hi-C技术是不是非常强大,做完GWAS没有鉴定到位于编码区的候选SNP,或者全是基因沙漠区的候选SNP,如何开展下游机制研究呢?不妨试一试Hi-C测序。
参考文献:Baxter J S, Leavy O C, Dryden N H, et al. Capture Hi-C identifies putative target genes at 33 breast cancer risk loci[J]. Nature communications, 2018, 9(1): 1028.




 京公网安备 11011302003368号
京公网安备 11011302003368号