

中文题目:炎症性肠病(IBD)患者的肠道菌群结构与代谢活性
影响因子:14.2
发表时间:2018.12
研究单位:麻省理工学院 哈佛大学
背景介绍
炎症性肠病(IBD)是一种由肠道微生物与肠道免疫系统相互作用异常而引发的慢性胃肠道炎症。IBD包括溃疡性结肠炎(UC)和克罗恩病(CD)两种主要亚型,它们分别分布于大肠和小肠,且拥有各自独特的微生物特征。研究表明,IBD患者的肠道微生物发生了巨大变化,与此同时,肠道微生物的代谢产物也能通过信号、免疫以及抗生素活性的途径改变结肠环境。然而,肠道特异微生物与它们的分子产物如何通过相互作用来触发、维持、减缓或预测IBD等炎症状况,我们目前仍不清楚。
短链脂肪酸(SCFAs)(如丁酸盐、乙酸盐和丙酸盐)是肠道微生物分解膳食纤维产生的小分子化合物,它能通过调节组蛋白去乙酰化酶抑制活性、基因表达、细胞增殖和免疫应答来影响宿主细胞。研究表明,IBD患者粪便中的丁酸盐含量降低,而丁酸盐可以通过调节Treg细胞的产生和增强巨噬细胞的活性预防结肠炎。共生微生物可以通过修饰代谢物改变宿主产生的信号分子。研究表明,肠道微生物产生的色氨酸脱氢酶能够将饮食中的色氨酸转化为色胺和其他分子。在CD患者的肠道微生物中,色氨酸代谢基因有所下降;缺失IBD易感基因(CARD)动物的微生物色氨酸代谢发生了改变,导致宿主更易患结肠炎。
肠道微生物的16S扩增子测序与代谢组学研究表明,IBD患者的代谢物发生了变化且菌群多样性普遍低于健康人群,与儿科IBD相关的肠道菌群也与代谢类型高度相关,粪便菌群结构与非靶向代谢组的相关性比靶向代谢组高。总之,肠道代谢组中存在与炎症和IBD相关的特征不明代谢物,这些代谢产物可能是由微生物衍生或修饰,非靶向模式为未知代谢物的捕获提供了可能。
本研究采用非靶向的液相色谱串联质谱(LC-MS)技术和宏基因组技术,研究了IBD患者粪便样品中的代谢组份和微生物的结构与功能。
研究方法
1. 样本收集(粪便):①PRISM(麻省总院IBD前瞻注册试验):CD患者(68例),UC患者(53例),健康人群组(34例)(Fig. 1a);②LifeLines DEEP和NLIBD(荷兰北部人群前瞻研究,用于人群疾病的验证):CD患者(20例),UC患者(23例),荷兰北部健康人群(22例)(图1.a)。
2. 宏基因组学:①DNA提取试剂盒:QIAamp DNA Stool Mini Kit (QIAGEN);②宏基因组建库测序:Illumina Hiseq 2500,2.5G数据/样品;③物种鉴定与功能注释:MetaPhlAn2,HUMAnN2。
3. 代谢组学:4种LC-MS模式。
● 质谱正离子模式(极性代谢物,如:有机酸):10μl匀浆,90μl萃取液(乙腈/甲醇/甲酸74.9:24.9:0.2,v/v)),内标(L-缬氨酸-d8和L-苯丙氨酸-d8),二氧化硅HILIC色谱柱(Waters)(150×2mm),流动相A(甲氨酸(10mM)和甲酸(0.1%)),流动相B(乙腈和甲酸(0.1%)),250µl /min流速,梯度洗脱(5%流动相A 保持1min,10min时提升至40%),全扫描分析模式(m/z:70-800,分辨率70000,获取速率3Hz),质谱参数(离子喷雾电压3.5kV,毛细管温度350℃,探头温度300℃,鞘气40,辅助气15,S-lens RF 40)。
● 质谱负离子模式(极性代谢物):30μl匀浆,120μl萃取液(80%甲醇),内标(肌苷15N4、胸腺嘧啶-D4和甘胆酸盐D4),Luna NH2色谱柱(Phenomenex),流动相A(乙酸铵(20mM)、铵水(20mM)),流动相B(氨水(10mM)/乙腈/甲醇(75/25),v/v),梯度洗脱(10%流动相A,至10min时至100%流动相A),全扫描分析模式(m/z:60-750,其余参数同上),质谱参数(探头温度325℃,鞘气55,其余参数同上)。
● 质谱负离子模式(中极性代谢物,如:胆酸、自由脂肪酸):30μl匀浆,90μl萃取液(甲醇),内标(E2-d4),10μl进样量,HSS T3色谱柱(Waters),流动相A(0.1%甲酸,水),流动相B(0.1%甲酸,乙腈),400µl /min流速,梯度洗脱(25%流动相A保存1min,至11min时至100%流动相B),全扫描分析模式(m/z:200-550,其余参数同上),质谱参数(探头温度300℃,毛细管温度320℃,鞘气45,辅助气10,S-lens RF 60)。
● 极性和非极性脂肪酸:10μl匀浆,190μl萃取液(异丙醇),内标(1-十二烷基-十三酰-丙三醇-3胆碱磷酸),10μl进样量,BEH C8色谱柱(Waters),流动相A(醋酸铵(10mM)/甲醇/甲酸95:5:0.1,v/v/v),流动相B(甲醇/乙酸99.9:0.1,v/v),450µl /min流速,梯度洗脱(80%流动相A保存1min,至2min时至80%流动相B,至7min时至100%流动相B并保持至10min),全扫描分析模式(m/z:200-1100,其余参数同上),质谱参数(探头温度300℃,毛细管温度300℃,鞘气50,辅助气15,S-lens RF 60)。
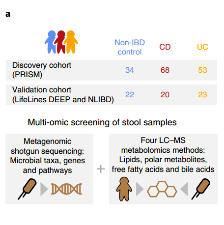
(图1.a:样本来源、类型及分析方法)
研究结果
本研究从155例(PRISM)样本中共检测出8,000多种代谢物,这些代谢物在健康人群与CD患者之间差异较大,UC患者的代谢物与健康人群和CD患者均有一定程度相似(图1.b)。粪便样品中的钙保护素与代谢物、菌群结构变化的第一主轴(PCoA)相关性均较强(图1.d.1e),UC患者的炎症程度既与健康人群相似又与CD患者相似,表明UC患者之间的肠道菌群结构与代谢组份差异较大。
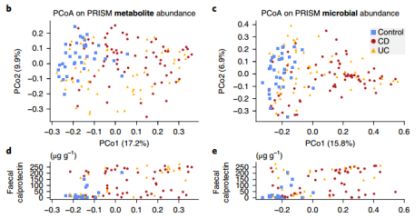
(图1.b-e:IBD与个体肠道微生物和代谢物的关联性)
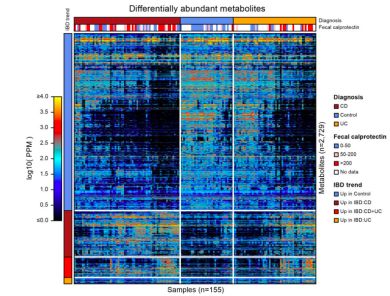
(图2:IBD代谢物丰度热图,图中显示了2729种IBD(UC/CD)与健康组之间具有丰度差异的特征代谢物)
2. IBD患者肠道代谢物的比较分析
本研究采用Benjamini-Hochberg FDR(q<0.05)方法进行多重检验校正,在IBD患者检测2729种丰度显著差异的代谢物,其中1931种在IBD患者中显著减少,224种在IBD患者中的显著增多,505种在CD患者中明显升高,仅69种只在UC患者中明显增多(与图1.b坐标轴中显示的结果一致)(图2)。基于HMDB注释定义了代谢物类并重点研究了97类,其中8类显著富集于CD患者,鞘脂类、甲亚胺酸和胆汁酸影响最大(图3.a);7类显著富集于UC患者,苯乙酸酯含量略有升高(图3.b)。IBD患者富集的胆酸包括胆酯(q=0.003)和鹅脱氧胆酸盐(q=0.0002)(图3.c),CD患者的次生胆汁酸(胆酸钠和脱氧胆酸)发生了补充消耗(q=0.06,0.13),鞘磷脂和神经酰胺在CD患者和UC患者中均明显过多(q<0.02)。多数代谢物类在IBD患者(CD和UC)中显著减少(图3.a.3.b),它们包括三萜、长链脂肪酸、苯基苯并二恶烷、胆固醇和三酰基甘油(C54:6)(图3.e.3.f)。另外,乳酸酯和泛酸酯(分子类无显著差异)在IBD患者中分别显著富集和减少(图3.g.3.h)。
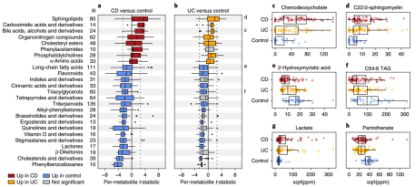
(图3. IBD患者和健康人群的肠道代谢物富集比较)
3. IBD患者的肠道代谢物模型
为了探究IBD患者肠道代谢物的生物模式,本文采用线性模型方法对2729种差异代谢物进行相似性聚类分析。结果表明,IBD患者“最大代谢物聚类簇”包含39种代谢产物,该聚类簇包含12种胆酸(推测)、17种未标记的代谢物、胆酯(图4.a浅绿色三角形)以及鹅脱氧胆酸酯(图4.a 深绿色三角形);健康人群的“最大代谢物聚类簇”包含62种高丰度代谢分子,四吡咯和衍生物均显著富集(图4.b)。
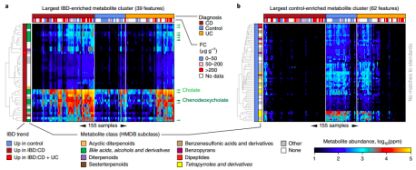
(图4. 基于丰度相关性的IBD患者代谢物聚类。FC,粪便钙防御蛋白)
4. IBD患者肠道菌群的“种”水平变化
基于Bray-Curtis算法的距离矩阵(PCoA)发现,CD患者与健康人群肠道菌群结构差异较大,而UC患者与前两者之间并无明显的区别,UC患者组内分布也较零散(图1.c.1.e)。为了进一步探究,本文运用线性建模研究195个“种”水平微生物。在IBD患者与健康人群之间发现了50种差异丰度微生物,其中35种在IBD患者中丰度较高,Roseburia hominis、Dorea formicigenerans和Ruminococcus obeum在健康人群中富集最多。与很多IBD研究报道一致,IBD患者的微生物种类明显减少,多样性普遍降低。“种”水平未分类的Roseburia(氏菌属)在UC和CD患者中显著富集,Bifidobacterium breve和Clostridium symbiosum仅富集于UC患者,CD患者具有20种独特且丰富的菌(如R. gnavus、Escherichia coli和Clostridium clostridioforme)(图5)。
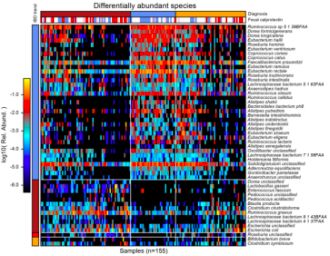
(图5. IBD患者“种”水平的菌群结构)
5. IBD相关微生物与代谢物的关联机制
微生物与代谢物之间的正相关性反映了两者之间的协同效应,为了确定这种关系,本研究对具有代表性的差异丰度菌种和代谢物进行关联分析(线性模型),发现了15,679例FDR(q<0.05)关联,其中乳酸与Pediococcus acidilactici呈正相关(r= 0.23,Spearman)。经过严格筛选,最终得到2279例关联(图6),其中包括122例标准和特征菌“种”的关联(图7.a),901个代谢物聚类簇,1878个丰度差异代谢物。然而,这些关联仅有6%是具有统计学意义且在对照组(健康人群)中被证实的。ETA(廿碳三烯酸)和DPA(二十二碳五烯酸,与CD相关的代谢物)与对照组相关的菌种呈负相关(Eubacterium ventriosum),与IBD相关的菌种呈正相关(R. gnavus)(图7.b.7.c)。CA(辛酸)对照组中高度关联的厌氧菌(Alistipes shahii、Alistipes putredinis和Alistipes finegoldii)正相关,与R. gnavus显著负相关(图7.d)。
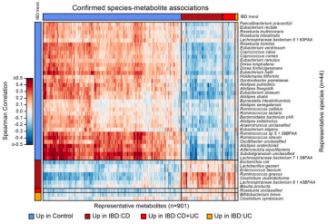
(图6.)
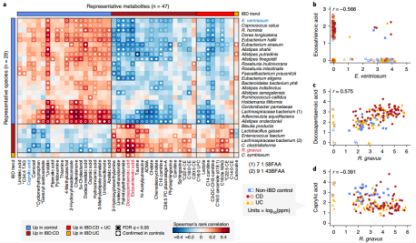
(图7. IBD相关微生物和代谢物的潜在关联)
6. IBD代谢相关微生物的功能
基于线性模型分析IBD微生物组的酶功能(EM)。与对照组相比,568种酶的丰度在UC或CD患者中差异显著(FDR,q<0.05)(图8.),其中246种酶不受任何单一菌种支配。UC和CD患者均富集有镁离子转运ATP酶(EC 3.6.3.2)(图9.a)和乙醇胺氨裂合酶(EC 4.3.1.7)(相对于对照组)(图9.b),而乙醇胺氨裂合酶与甘油磷脂的合成有关,后者在UC和CD患者的代谢物中富集最多(图3.a)。谷胱甘肽还原酶(EC 1.8.1.7)在IBD患者中也有较高丰度(图9.c);丙酮酸合成酶(EC 1.2.7.1)在对照组中富集,在CD患者中完全未检测到(图9.d);维生素B12(如前咕啉,EC 1.3.1.76)在对照组含量较高(图9.e)。四吡咯可能是维生素B12的衍生物和相关化合物,它在LC-MS代谢组分析中普遍存在,并且在IBD患者中富集(图4.b)。
“代谢组-基因功能(酶)”关联模型遵循“菌种-基因功能(酶)”关联,多数代谢物仅与少数基因功能(酶)存在小部分(3%)关联(反之亦然),而多数(95%)“代谢组-基因功能(酶)”关联与IBD的发病机理有关。这些小部分“代谢组-基因功能(酶)”关联中,镁离子转运ATP酶与羟基十四烷酸显著负相关(r=-0.492,Spearman)(图9.f),precorrin-2脱氢酶与正已酸正相关(r=0.507)(图9.g)。
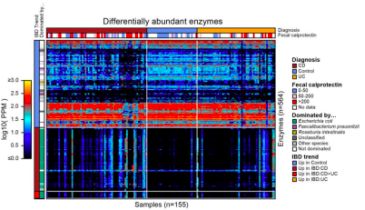
(图8.)
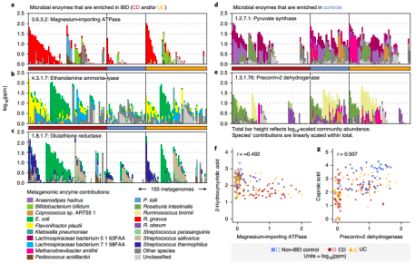
(图9. IBD代谢相关微生物的功能)
7. Multi-omic特征区分群组中IBD子集
最后,本研究从代谢物、微生物菌“种”及两者组合水平训练“随机森林”(RF)分类器,鉴定IBD病症和IBD子集,受试者工作曲线(ROC)描绘RF分类器的假阳性率和准确率,混合矩阵评估PRISM队列IBD子集RF分类器。结果显示,所有RF分类器对IBD患者和健康人群的辨识效果良好,AUC值为0.86-0.92,交叉验证结果(AUC:0.90-0.92)更优于独立验证结果(AUC:0.86-0.89)(图10.a)。PRISM队列中,代谢物、微生物菌“种”及两者组合水平预测UC、CD和健康人群的准确率为64-65%,该结果虽然不如案例/对照成功,但仍高于随机预测(正确率:30%)(图10.b),将PRISM训练IBD子集RF分类器应用于荷兰人群时(用于人类疾病验证),输入数据类型之间差异较大(图10.b)。
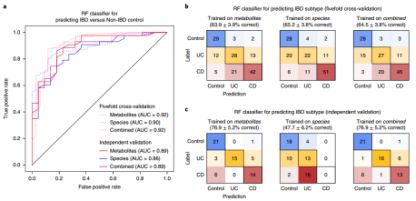
(图10.肠道微生物Multi-Omics analysis预测IBD病症及子集)
===其他相关研究(代谢组学+宏基因组学)===
1. 婴儿肠道微生物在发育和Ⅰ型糖尿病发展中的动态变化
Kostic A D, Gevers D, Siljander H, et al. The dynamics of the human infant gut microbiome in development and in progression toward typeⅠdiabetes [J]. Cell host & microbe, 2015, 17(2): 260-273.
2. 二甲双胍改变了治疗初期Ⅰ型糖尿病患者的肠道微生物,促进了该药的疗效
Wu H, Esteve E, Tremaroli V, et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug [J]. Nature medicine, 2017, 23(7): 850.
3. 药物治疗对心血管疾病模型(apo-E−/−)小鼠肠道微生物和代谢物的影响
Ryan P M, London L E E, Bjorndahl T C, et al. Microbiome and metabolome modifying effects of several cardiovascular disease interventions in apo-E−/− mice [J]. Microbiome, 2017, 5(1): 30.
如果您的项目有问题,欢迎咨询我们,我们将免费为您设计文章方案。





 京公网安备 11011302003368号
京公网安备 11011302003368号